Introduction
Dental findings in many genetic conditions constitute an important feature of the disorder. Therefore recognition of dental findings in syndromes make them indispensable in the diagnosis of the syndromic condition. Hypodondia i.e agenesis of one or more teeth is invariably reported with ectodermal dysplasia[1]. Hyperdontia or presence of supernumerary teeth is associated with cleidocranial dysostosis[2] and Gardner’s syndrome[3]. Root dilacerations is associated with certain syndromes and developmental disorders, such as the Smith-Magenis syndrome[4], the hypermobility type of Ehlers-Danlos syndrome[5], the Axenfeld-Rieger syndrome[6] and congenital ichthyosis[7]. In Ellis Van Crevald syndrome[8] crown morphology is altered where conically shaped lateral incisors and canines and barrel shaped central incisors are present.
In 1974 Nance et al[9] and Horan and Billson[10] first described a type of uncommon X linked congenital cataract with associated characteristic dental features and microcornea. This syndrome is referred as Nance-Horan syndrome (NHS) or the Cataract-Dental Syndrome(MIM No.: 302350).Van Dorp and Delleman[11] were able to confirm and broaden the clinical description of the ophthalmological and facial manifestations and Bixler et al[12] showed that the dental and ophthalmological features in female carriers could be more severe than previously recognized. The dental anomalies in NHS were described by Seow et al[13] in 1985.
Very few case reports have been reported in the dental literature. This article aims to provide a literature review of Nance Horan Syndrome (NHS) and present a case report with emphasis on associated dental abnormalities, few of which have not been previously reported like Dens In Dente, transmigration and talons cusp.
Differential Diagnosis
The differential diagnosis for NHS should include diseases like isolated cataract, X linked micropthalmia, Lenz syndrome oculo-cerebro-renal (Lowe) syndrome, Ectodermal dysplasia and Congenital syphilis.
Etiology And Incidence:
In Nance-Horan syndrome (NHS) affected males have congenital cataracts or micro cornea, and approximately 50% develop glaucoma. Characteristic dental anomalies are tapered screwdriver-shaped incisors and mulberry-like molars, with supernumeraries reported in 65% of cases[14]. NHS is transmitted as an X-linked disorder and molecular genetic analysis has refined the locus to a 3.5-cM interval on the distal short arm of the X chromosome, between Xp22.31 and Xp22.13.[15] This region also contains the amelogenin gene AMGX, and therefore it has been postulated that NHS may be a contiguous gene syndrome. Analysis has so far failed to demonstrate evidence of an AMGX gene deletion in NHS.[16] Focal Dermal Hypoplasia (DHOF), also termed Goltz syndrome, is a further disorder demonstrating dental anomalies that maps to this region, Xp22.31.[17] MIDAS syndrome (micropthalmia with linear skin defects) also maps to Xp22.3;severe micropthalmia, sclerocornea, cataracts, and glaucoma are all reported in this syndrome. It has been postulated that MIDAS and DHOF, together with Aicardi syndrome, may be due to involvement of the same gene, with different patterns of X-inactivation being responsible for the variations in phenotype.[18] Although it has also been suggested they may be a contiguous gene syndrome.[19] It is of much interest that different syndromes, with similar reported dental or ocular anomalies, have been mapped to the same cytogenetic location and further research is warranted in this area.
Another potential candidate gene, which flanks the NHS locus, is the human homologue of a mouse retinoic acid induced gene (RAI2). Retinoic acid and in particular the retinoic acid receptor a[COMP: lc alpha should have acute accent] gene (RARA) have been implicated in craniofacial malformations, especially cleft lip and palate.[20] It has not, however, been implicated in dental development. Direct sequencing of RAI2 in 9 NHS-affected families has been undertaken; however, no mutations were revealed and therefore it is unlikely RAI2 is associated with NHS.[21]
Clinical Features (As Described By Toutain[22]):
A. In Male Patients
1. Ocular findings
In cases with known family history the diagnosis is made early, generally in the first year of life, and most often at birth. The congenital cataract is present in 100% of cases, it is bilateral, usually severe, dense and most often total and is associated with microcornea (96% cases), or even microphthalmia. In the majority of cases (93%) it is responsible for severe visual impairment evidenced by nystagmus (93%), sometimes associated with strabismus (43%), and surgery is generally required (89% cases). Postoperative complications include glaucoma (28%), which is generally poorly controlled by medical treatment, retinal detachment (sometimes iterative) (14%), corneal lesions (14%), and even eyeball atrophy (12%) are frequent and severe and may necessitate enucleation. Even after surgery, visual prognosis remains poor overall.
2. Dental abnormalities
Dental abnormalities are of high diagnostic value and are present in 100% of cases. They are characteristic and specific of NHS, either by their type or by their aggregation in the same individual. Permanent and deciduous teeth are involved. Although typical and often marked, dental anomalies are easily overlooked. Their recognition requires careful physical and radiological dental examination.
a) Crown shape anomalies
Characteristic anomalies are observed on the incisors, described as screwdriver-shaped or conical, sometimes with a medial notch or an irregular incisal edge, or also called Hutchinson's teeth. Less typically, the incisors may be pointed, small, narrow or cylindrical. The existence of cingulate cusps is characteristic. The canines are often enlarged, globular, sometimes dome-shaped or bud-shaped, or with a trilobate edge. Premolars and molars are rounded, globular and sometimes small. The common presence of central supernumerary cusps gives them a mulberry or lotus flower shape.
b) Number anomalies
Number anomalies consist of supernumerary teeth (posterior teeth or incisors), which are often impacted. A characteristic finding is the mesiodens (median incisor located behind the normal upper incisors). Dental agenesis is possible (canines, premolars, molars). Impacted teeth are common, generally involving the supernumerary teeth, but they may also involve normal teeth (canines, molars).
c) Position and implantation anomalies
A diastema is found in all cases, most commonly present between the upper incisors. Malpositions or malimplantations (excluding common malpositions) consisting of germ translation or ectopic position, are possible.
d) Other anomalies
Late persistence of deciduous teeth, or pulp chamber anomalies (taurodontism, wide pulp chambers, abnormally calcified pulps, pulp stones) are observed with significantly higher frequency than in the general population.
3. Dysmorphic features
Facial dysmorphism is constant but sometimes subtle and difficult for an inexperienced examiner to recognize. It may include: -
Ø A long, sometimes narrow, often rectangular face
Ø Marked, long sometimes vertical chin, and prognathism in all cases
Ø A large nose, with a high, narrow nasal bridge
Ø Large, often protruding ears.
4. Mental retardation
Intellectual impairment is observed in about 30% cases. It is usually (80%) mild or moderate, homogeneous, without motor delay, but profound retardation is possible (20%) and is associated with autistic features.
Interfamilial phenotypic variability is observed but there is correlation in the severity of ocular, dental and dysmorphic features. Intellectual impairment also has marked inter- and interfamilial variability of expression but the existence and the degree of mental handicap are not associated with the severity of the other clinical findings.
B. In Heterozygous Female
Clinical manifestations are identical to those of affected males but they are attenuated and are often limited to subclinical findings.
1. Ocular signs
Ocular signs are not always present but they are observed in more than 90% cases.
They consist of bilateral, but often asymmetrical, predominantly posterior lens opacities. Extensive or progressive opacities are observed in 18% of the cases. Micro cornea is rare (6%) and microphthalmia has never been reported. Surgery is required in only a minority of cases (8%) with progressive lesions at an advanced age or with extensive congenital cataract. Postoperative complications do not usually occur and visual prognosis is good overall, vision generally being normal (>75% cases), or mildly decreased at an advanced age. Significantly low visual acuity is reported in only 3% of cases and strabismus in 2% of cases.
2. Dental Anomalies
Dental anomalies are present in all cases. They are identical to those observed in affected males but are generally less severe and less varied. They may be very subtle and are usually overlooked. In a few cases they are marked and require intensive orthodontic treatment.
3. Facial Appearance
Facial appearance resembles that of affected males, but dysmorphic features are inconstant, often subtle and difficult for a non-trained examiner to recognize.
4. Intellectual Impairment
There is no intellectual impairment in heterozygotes. expression of clinical signs in heterozygotes is not related to X chromosome inactivation.
Management Including Treatments
1. Ocular abnormalities- They usually require surgery for cataract extraction although the results are poor. Complications (glaucoma, retinal detachment) are treated medically or surgically depending on the type and severity. The ocular problem requires education appropriate for the degree of visual handicap and often necessitates education in special school for the visually impaired.
2. Dental anomalies- Require orthodontic treatment for aesthetic reasons.
3. Intellectual impairment- Requires special education.
Genetic Counseling
In the offspring of an affected male, all the daughters are heterozygotes and all the sons are unaffected. A heterozygous female has a 50% chance of transmitting the mutated gene: she has a 25% chance of having an affected son and a 25% chance of having a carrier daughter.
Heterozygote detection is based on the recognition of physical ocular and/or dental signs, which are generally subclinical, and requires a directed ophthalmological examination and careful and complete clinical and radiological dental examination performed by an experienced examiner. Indirect molecular study based on the analysis of markers linked to the gene may be performed in familial cases, in comparison with the clinical data.
Prenatal diagnosis
It is possible by indirect molecular analysis in informative families.
Case Report:
A 13-year-old male was referred to the Orthodontic Department for the management of supernumerary teeth and impacted teeth.
The provisional diagnosis of the Nance-Horan syndrome (NHS) was made owing to the presence of triad of bilateral congenital cataracts in association with the distinctive dental findings and characteristic facial dysmorphism.
Clinical Features
1. Opthalmologic Findings:
This patient was born with bilateral congenital cataracts and had undergone surgery for removal of the right-sided cataract. The family had never been offered an etiology for the cataracts and had not been investigated for the presence of a syndrome. (Fig. 1).
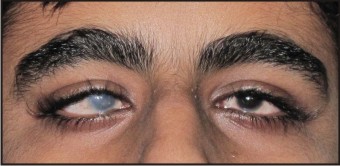 | Fig .1 : Figure Showing The Ophthalmologic Abnormalities.
 |
2. Dental Features:
The patient had a well maintained dentition, with good oral hygiene and gingival health. No restorations or carious lesions were present. The Dental anomalies found were:
(A) Crown and root shape abnormalities:
1) Hutchinsonian incisor: A medial notch or an irregular incisal edge of maxillary central incisors.(Fig. 2).
2) Theupper incisor teethwere pointed, cylindrical, thin, tapering, and screwdriver-shaped. (Fig. 3).
3) Talons cusp or cingulated cusp:The existence of cingulate cusps is characteristic.Premolars and molars are rounded, globular and small. The common presence of central supernumerary cusps gives them a mulberry or lotus flower. (Fig. 3).
4) Dens in Dente: Dens invaginatus, also known as dens in dente ("tooth within a tooth") is a condition found in teeth where the outer surface folds inward. Dens invaginatus is a malformation of teeth most likely resulting from an infolding of the dental papilla during tooth development or invagination of all layer of the enamel organ in dental papillae.
In the patient dens in dente was seen in Mandibular right canine.(Fig. 4).
5) Dilacerated crowns and roots: The abnormal angulation’s of crown is seen with respect to roots in maxillary central incisors. Dilacerated roots are also noted in several teeth (Fig. 5).
(B) Tooth Position and implantation anomalies :
Transmigration of tooth is seen in mandibular anterior segment as shown in the CBCT section .Transposition of canine and premolar is seen in the right maxillary segment. 13 is impacted. (Fig. 5).
(C) Tooth number anomalies:
A characteristic finding is the mesiodens (median incisor located behind the normal upper incisors). In this case two mesiodens are present in the region of 11 & 21. Supernumerary teeth are present in the region of 26, 45& 46. Horizontally impacted and transmigrated supernumerary are noted in the basal bone lying one above the other. Supernumerary teeth are also present with respect to 31, 33, 34 and 36.Paramolars are present with respect to 17, 27, 37 & 47. (Fig. 6 a, b).
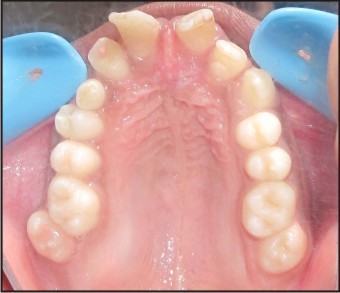 | Fig .2 : Inraoral Occlusal View Showing Talons Cusp And Globular Shaped Molars And Hutchinsonian Incisors.
|
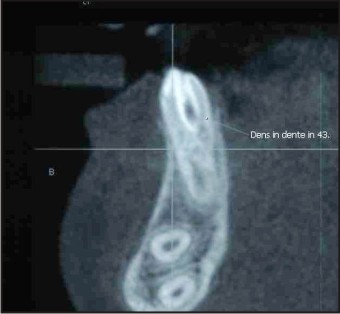 | Fig .3 : Cbct Section Showing Dens In Dente.
|
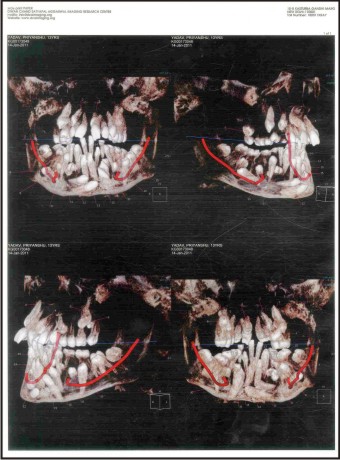 | Fig .4 : Cbct Sections Showing Various Supernumerary, Para Molars And Dilacerated Roots And Crowns, Transmigration, Transposition
|
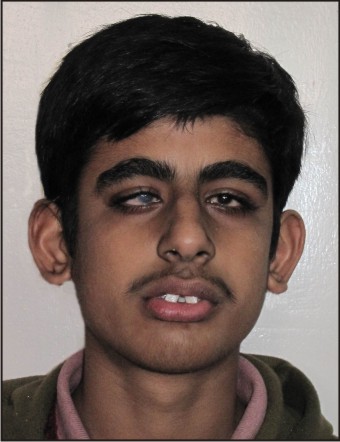 | Fig .5 : extra Oral Frontal View Showing Facial Dysmorphism And Anteverted Pinnae.
|
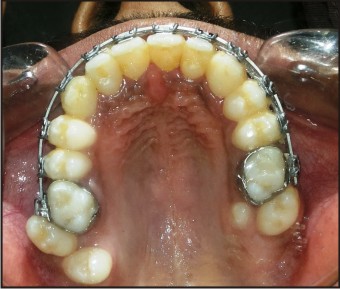 | Fig .6 (A) : Active Orthodontic Treatment.
|
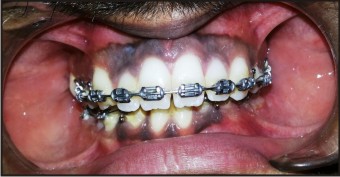 | Fig .6 (B) : Active Orthodontic Treatment.
|
Facial dysmorphism:
Patient had a prognathic, long narrow face, vertical chin, and a large nose, with high, narrow nasal bridge. The ears were long and prominent with anteverted pinnae.
Differential diagnosis:
The presence of thin tapering teeth with anterior spacing was suggestive of a form of ectodermal dysplasia; however, a complete permanent dentition (with the exception of third molars) and the presence of supernumerary teeth excluded this diagnosis. ‘‘Screwdriver-shaped’’ incisor teeth and mulberry like molars are a commonly reported feature of congenital syphilis; there were, however, no other general features to support this diagnosis.
Management and treatment considerations:
(1) Counselling Those families so far described with Nance-Horan syndrome most strongly support X linked recessive inheritance. Firstly, manifestations in the heterozygous female have been less marked than in the hemizygous male. There has been no example of male to male transmission.
(2) Ocular abnormalities .patient had undergone surgery for removal of right sided cataract and is under constant supervision for further treatment.
(3) Dental abnormalities: Selected supernumerary teeth have been extracted .Patient is undergoing active orthodontic treatment.
Conclusion:
Dentofacial features are often the most obvious sign of an underlying syndrome, which may have important implications for the health of the patient and their potential future offspring. Management will often require the involvement of medical specialties, in particular clinical Genetics, Ophthalmologist and dental specialties mainly Oral pathologist, Oral surgeons, Orthodontist and Prosthodontist.
References:
1. Lowry RB, Robinson GC, Miller JR: Hereditary ectodermal dysplasia. Symptoms, inheritance patterns, differential diagnosis, management. Clin Pediatr 1966; 5:395-402.
2. Kalliala E, Taskinen PJ: Cleidocranial dysostosis. A report of six typical cases and one atypical case. Oral Surg 1962; 15:808-22.
3. Gardner EJ, Richards RC: Multiple cutaneous and subcutaneous lesions occurring simultaneously with hereditary polyposis and osteomatosis. Am J Hum Genet, 1953 5:139-47.
4. Tomona N, Smith AC, Guadagnini JP, Hart TC. Craniofacial and dental phenotype of Smith-Magenis syndrome. Am J Med Genet A 2006; 140:2556–61.
5. Yassin OM, Rihani FB. Multiple developmental dental anomalies and hypermobility type Ehlers-Danlos syndrome. J Clin Pediatr Dent 2006; 30:337–41.
6. Jena AK, Kharbanda OP. Axenfeld-Rieger syndrome: report on dental and craniofacial findings. J Clin Pediatr Dent 2005;30:83–8.
7. Pinkham JR, Casamassimo PS, McTigue DJ, Fields HW,Nowak AJ. Pediatric dentistry: infancy through adolescence. St Louis: Elsevier Inc; 2005. p. 66.
8. Feingold M, Jankoski J, Johnson D, Darling DB, Kreidberg MB, Wilson O, Cohen MM, Gellis SS: Ellis-van Crevald syndrome. Clin Pediatr , 1966; 5:431-36.
9. Nance WE, Warburg M, Bixler D, Helveston EM. Congenital Xlinked cataract, dental anomalies and brachymetacarpalia. Birth Defects 1974;X(4):285-91.
10. Horan MB, Billson FA. X-linked cataract and Hutchinsonian teeth. Aust PaediatrJ 1974;1O:98-102.
11. Van Dorp DB, Delleman JW. A family with X-chromosomal recessive congenital cataract, microphthalmia, a peculiar form of the ear and dental anomalies. J Pediatr Ophthalmol Strabismus 1979;16: 166-71.
12. Bixler D, Higgens M, Hartsfield J Jr. The Nance-Horan syndrome: a rare X-linked ocular dental trait with expression in heterozygous females. Clin Genet 1984;26:30-5 .
13. Seow WK, Brown JP, Romaniuk K. The Nance-Horan syndrome of dental anomalies, congenital cataracts micropthalmia and anteverted pinna: case report. Paediatr Dent 1985;7:7-11.
14. Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the head and neck. 4th ed. New York: Oxford University Press; 2001. p. 1108.
15. Toutain A, Ronce N, Dessay B, et al. Nance-Horan syndrome: linkage analysis in 4 families refines localization in Xp22.31ep22.13 region. Hum Genet 1997;99:256-61.
16. Franco E, Hodgson S, Lench N, Roberts GJ. Nance-Horan syndrome: a contiguous gene syndrome involving deletion of the amelogenin gene? A case report and molecular analysis. Oral Dis 1995;1:8-11.
17. Naritomi K, Izumikawa Y, Nagataki S, et al. Combined Goltz and Aicardi syndromes in a terminal Xp deletion: are they a contiguous gene syndrome? Am J Med Genet 1992;43:839-43.
18. Lindsay EA, Grillo A, Ferrero GB, et al. Micropthalmia with linear skin defects (MLS) syndrome: clinical, cytogenetic and molecular characterization. Am J Med Genet 1994;49:229-34.
19. Mu¨cke J, Happle R, Theile H. MIDAS syndrome respectively MLS syndrome: separate entity rather than a particular lyonization pattern of the gene causing Goltz syndrome. Am J Med Genet 1995;57:117-8.
20. Juriloff DM, Mah DG. The major locus for multifactorial nonsyndromic cleft lip maps to mouse chromosome 11. Mamm Genome 1995;6:65-9.
21. Walpole SM, Ronce N, Grayson C, et al. Exclusion of RAI2 as the causative gene for Nance-Horan syndrome. Hum Genet 1999;104:410-1.
22. Toutain A Nance –Horan Syndrome .Orphanet encyclopaedia June 2003.
|