Introduction:
Joubert syndrome (JS) is an autosomal recessive disorder characterized by hypotonia in infancy with later development of ataxia, abnormal breathing pattern, abnormal eye movements, development delays and mental retardation. It arises due to the hypoplasia of cerebellar vermis and brainstem malformation[1],[2],[3]. As the cerebellum governs movement and balance, the patients of Joubert syndrome suffer from hypotonia and ataxia. Other variable features that are associated with this syndrome include nephronophthisis, retinal dystrophy, ocular coloboma, occipital encephalocele, polydactyly, hepatic fibrosis and endocrine abnormalities. The multitude of signs associated with Joubert syndrome has given rise to a broader nomenclature called Joubert syndrome related disorders (JSRD)[4].
Recently, based on the systemic multiorgan involvement, six phenotypes of the JSRD spectrum have been described[5]: 1) "pure" JS (purely neurological without retinal, renal, or liver involvement); 2) JS with ocular defect (neurological features associated with retinal dystrophy); 3) JS with renal defect (neurological features with renal involvement, mostly nephronophthisis); 4) JS with oculorenal defects (association of neurological signs with both retinal dystrophy and nephronophthisis); 5) JS with hepatic defect (neurological features with congenital liver fibrosis); and 6) JS with oral-facial-digital defects.
A review of literature reveals that case reports of Joubert syndrome and related disorders are few and the oral-facial-digit subtype of this rare neurological condition is of particular interest to the dental professionals.
Clinical Case Presentation:
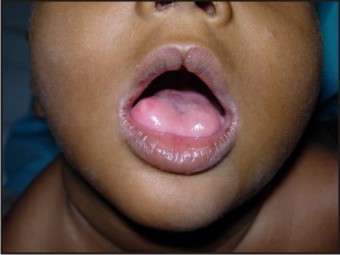
A three and a half year old girl reported to the private practice with the chief complaint of swelling on right side of tongue since birth and multiple carious teeth. The swelling was not painful, and did not cause any discomfort during mastication. The child’s parents gave a history of consanguineous marriage. The delivery was normal and at full term. (Fig 1)
The child’s medical history revealed:
- Enlarged head since birth. She was diagnosed as suffering from congenital hydrocephalus
- Breathlessness in early infancy which disappeared at the age of one and half years
- Delayed milestones
- Mental retardation
- Speech delay
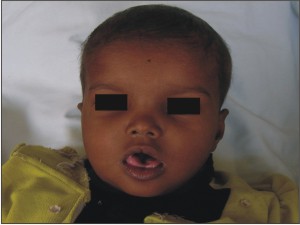 | Fig 1: Pre-operative Extra Oral Photograph Of Face
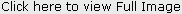 |
On general examination, the following findings were noted:
- The patient had frontal bossing, hypertelorism, flat nasal bridge and lack of head holding.
- She displayed signs of generalized hypotonia and ataxia. She could not sit, stand or walk without support
- The patient was found to be mentally challenged. She could not follow any instructions given to her
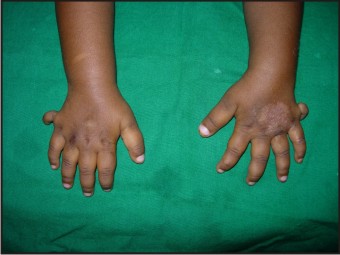
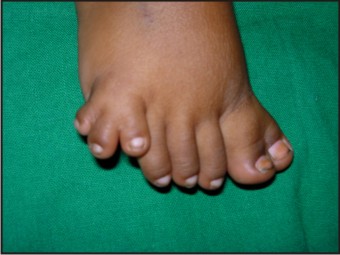
- The patient had post axial polydactyly affecting all four limbs and pre axial polysyndactyly in both feet (Fig 2, 3 & 4) Complete intraoral examination revealed, (Fig 5, 6).
 | Fig 2: Post Axial Polydactyly Present In Both Hands
|
 | Fig 3: Post Axial Polydactyly With Pre Axial Polysyndactyly In Feet
|
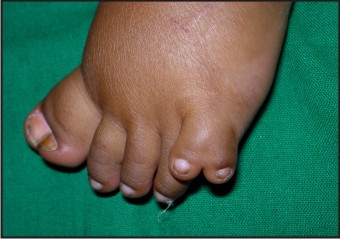 | Fig 4: Post Axial Polydactyly With Pre Axial Polysyndactyly In Feet
|
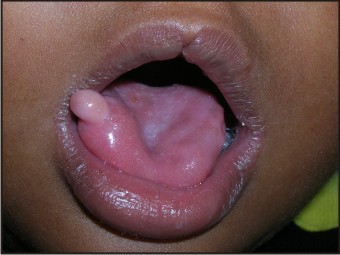 | Fig 5: Intraoral Photograph Showing Notched Upper Lip And Swelling On Tongue
|
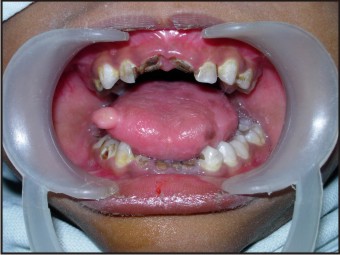 | Fig 6: Intraoral Photograph Showing Carious Teeth
|
- Central notching of vermillion border of upper lip
- An enlarged and protruded tongue
- An oval, pink, firm swelling measuring 5 mm (approximately) in diameter present on the right lateral border of the anterior 2/3rds of the tongue. The lesion was sessile, not tender and had a smooth surface.
- The oral hygiene status was poor with root fragments in relation to 51, 61, 71, 72, 81 and 82. Tooth no. 54 was grossly decayed and fractured. Teeth no. 52, 54, 62, 63, 64, 73, 83, 84 and 85 were decayed.
The patient was thoroughly investigated for multisystem involvement. Based on the complaints and extra and intra oral findings, the following investigations were carried out,
- Skull radiographs: Posteroanterior and lateral skull views were taken which showed enlarged skull and confirmed the hydrocephalus
- MRI scan of head revealed the classic ‘Molar tooth sign’ (MTS). It is the appearance caused due to the hypoplasia of the cerebellar vermis and the accompanying brain stem abnormalities which resemble the section of a molar tooth (Fig 7).
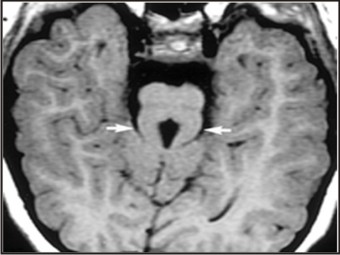 | Fig 7: Axial Mri Image Showing Molar Tooth Sign (Mts)
|
- Radiographs of the hands and feet were taken which confirmed the extra digits along with the underlying bone (Fig 8, 9) - Chest radiograph revealed normal lung parenchyma
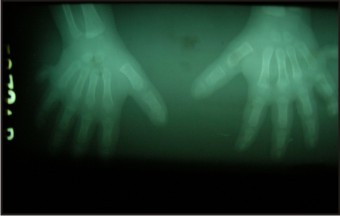 | Fig 8: Radiographs Of Hands
|
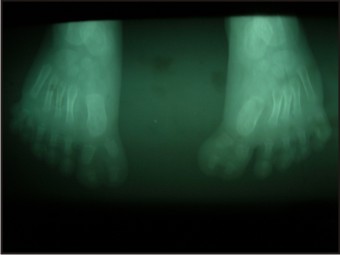 | Fig 9: Radiographs Of Feet
|
- Routine blood and urine analysis was found to be within normal limits.
A provisional diagnosis of Joubert syndrome was reached on the account of: hypotonia and ataxia, Molar tooth sign on CT scan, developmental delays,hypertrophy of tongue along with the presence of fibroma on the lateral border of the tongue, polydactyly of all limbs, syndactyly of toes.
The ‘molar tooth sign’ (MTS) seen in CT scan images in Joubert syndrome is also seen in a few other conditions such as COACH syndrome (Cerebellar vermis hypoplasia, Oligophrenia, Ataxia, Coloboma, Hepatic fibrosis)[6],[7] Senior-Loken syndrome (retinopathy, juvenile onset nephronophthisis)[8],[9] and Dekaban-Arima syndrome (cystic dysplastic kidneys)[10]. In the present case, ophthalmologic and nephrologic investigations were non contributory. Some individuals exhibiting molar tooth sign also have sever retinal dysplasia with congenital blindness resembling Leber congenital amaurosis.
Differentiation should also be made from Dandy Walker malformation which is a hindbrain malformation characterized by hypoplasia of the cerebellar vermis and/or the cerebellar hemispheres. This condition also involves an enlarged retrocerebellar cerebrospinal fluid collection that is continuous with the fouth ventricle (often misnamed as ‘cyst’ of posterior fossa). Associated findings in some individuals include agenesis or hypoplasia of the corpus callosum and hydrocephalus[11]. MRI findings are helpful in distinguishing Dandy Walker malformation from the molar tooth sign[12].
The treatment was planned to be carried out under general anesthesia after obtaining fitness from the child’s neurologist the anesthetists. An excisional biopsy of the lesion on the tongue was planned. The lesion was excised followed by placement of suture for hemostasis. The specimen was sent to pathology laboratory for histopathological examination. Root fragments in 51, 61, 71, 72, 81, 82 region were extracted along with tooth 54. One suture was placed in relation to 51, 61 extraction sockets and another suture in extraction socket of 54. Teeth no. 52, 62, 63, 64, 73, 84, 85 were restored using glass ionomer cement.
Histologically, the excised lesion showed parakeratinized, stratified squamous epithelium with elongated rete pegs. The connective tissue showed dense fibrous tissue, blood vessels, muscle fibres and a few salivary acini. The overall histopathological pattern was suggestive of irritational fibroma (Fig 10). On recall appointment, satisfactory healing of the excision wound and extraction sockets was noted (Fig 11). The patient was referred for genetic testing and a six monthly recall was advised.
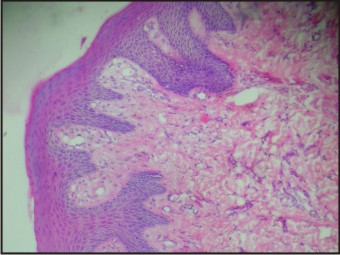 | Fig 10: Histopathological Image
|
 | Fig 11: Post Operative Intraoral Photograph
|
Discussion
Joubert syndrome was first reported by a French neurologist, Marie Joubert in the year 1969[1]. She described the condition in four siblings with agenesis of the cerebellar vermis, episodic hyperpnea, abnormal eye movements, ataxia and intellectual disability. Flannery et al[13] found the prevalence of Joubert syndrome to be 1:100,000-1:150,000. A male preponderance ratio of 1:2 over females has been reported[14].
A few years later, researchers reported the presence of a pathognomonic midbrain-hindbrain malformation called the ‘Molar tooth sign’ (MTS) in cranial MRI images of patients with Joubert syndrome[15]. The presence of MTS was also reported in few other disorders previously considered as distinct entities, hence the term Joubert syndrome related disorders was coined to group all conditions sharing the MTS[4],[5]. MTS is the appearance given by hypoplasia of the cerebellar vermis and accompanying brainstem abnormalities on axial imaging through the junction of the midbrain and pons (isthmus region).[15],[16] The resulting images resemble a tooth. The molar tooth sign comprises an abnormally deep interpeduncular fossa; prominent , straight and thickened superior cerebellar peduncles and hypoplasia of primarily the superior vermis found at the midline of the cerebellum. The absence of a normal vermis creates a midline cleft between the two cerebellar hemispheres, which results in a characteristic ‘bat wing’ appearance of the fourth ventricle on transverse CT and MR images.
The cluster of other signs and symptoms seen in Joubert syndrome related disorder has variable presentation. Authors of case reviews of patients with JSRD have reported the prevalence of some of the associated findings as polydactyly (8%), ocular coloboma (4%), and hamartoma of tongue (2%).[17],[18] Many clinical features of Joubert syndrome are evident in infancy. Most children suffering from Joubert syndrome exhibit truncal ataxia and delayed motor milestones. Many also have rhythmic tongue movements which may lead to tongue hypertrophy and tumours. Surgical reduction of the tongue has been suggested only if the enlarged tongue interferes in speech, mastication or obstructs respiration[1],[19].
Many children with JSRD demonstrate horizontal nystagmus at birth, which improves with age. Ocular colobomas can occur in the iris, choroid, or retina[19].
The most common manifestation of renal involvement in JSRD is cystic dysplasia which on renal ultrasound is visualized as multiple small cysts in immature kidneys with fetal lobulations[14],[20]. Glomerulosclerosis and fibrotic changes of the kidneys have also been described. In general, renal involvement is rarely present at birth and requires monitoring since it can develop during childhood and adolescence[20].
Hepatic fibrosis has also been described in some children suffering from JSRD and is often associated with renal cystic dysplasia[21]. The present case had no signs of hepatic or renal involvement. Polydactyly can be unilateral or bilateral and is often postacial, although preaxial polydactyly of the toes is also frequently reported as is seen in the present case[4],[22].
Typical facial features seen in JSRD include high arched eyebrows, broad nasal bridge with anteverted nostrils, triangular shaped mouth, and low-set ears have been described. Most of these facial features were seen in the present case.
Considering the wide range of clinical presentations, prenatal diagnosis by means of ultrasound, CT and MRI scanning of women with a family background of JSRD gains special importance. With the help of MRI scans, the diagnosis of JSRD can be made prenatally before 24 weeks’ gestation[23].
Initial prognosis is related to the extent and severity of breathing dysregulation. In particular, recurrent episodes of prolonged apneas can be life-threatening and require assisted ventilation. In most cases, these respiratory abnormalities resolve spontaneously in the first few months of life[5]. After infancy, prognosis depends mostly on renal and hepatic complications that, if not timely diagnosed and managed, represent the major causes of death in JSRD patients.
Conclusion
The dental management of children with systemic disorders and syndromes is challenging and tends to be a multidisciplinary one. Periodic recall and follow up care of such patients goes a long way in providing comprehensive care and improving the quality of life.
References
1. Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 1969; 19: 813–825.
2. Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol 1999; 14: 583–590; discussion 90–91.
3. Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 1997; 12: 423–430.
4. Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, et al. Molar tooth sign of the midbrain–hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A 2004; 125A:125–134.
5. Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20.
6. Verloes A, Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am J Med Genet. 1989 Feb;32(2):227-232.
7. Gentile M, Di Carlo A, Susca F, Gambotto A, Caruso ML, Panella C, et al. COACH syndrome: report of two brothers with congenital hepatic fibrosis, cerebellar vermis hypoplasia, oligophrenia, ataxia, and mental retardation. Am J Med Genet. 1996 Aug 23;64(3):514-520.
8. Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapetoretinal degeneration. A new oculorenal dystrophy. Am J Ophthalmol. 1961 Nov;52:625-633.
9. Loken AC, Hanssen O, Halvorsen S, Jolster NJ. Hereditary renal dysplasia and blindness. Acta Paediatr. 1961 Mar;50:177-184.
10. Dekaban AS. Hereditary syndrome of congenital retinal blindness (Leber), polycystic kidneys and maldevelopment of the brain.Am J Ophthalmol. 1969 Dec;68(6):1029-1037.
11. Patel S, Barkovich AJ. Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol. 2002 Aug;23(7):1074-1087.
12. Maria BL, Bozorgmanesh A, Kimmel KN, Theriaque D, Quisling RG. Quantitiativve assessment of brainstem development in Joubert Syndrome and Dandy-Walker syndrome. J Child Neurol 2001;16:751-758.
13. Flannery DB and Hudson JG. A survey of Joubert syndrome. David W Smith workshop. Prc Greenwood Genet Ctr. 1994;13:130.
14. Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet 1992;43:726-731.
15. Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, et al “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 1997;12:423-430.
16. Quisling RG, Barkovich AJ, Maria BL. Magnetic resonance imaging features and classification of central nervous system malformations in Joubert syndrome. J Child Neurol 1999;14:628-635.
17. Chance PF, Cavalier L, Satran D, Pellegrino JE, Koenig M, Dobyns WB. Clinical nosologic and genetic aspects of Joubert syndrome and related syndromes. J Child Neurol 1999;14:660-666.
18. Sztriha L, Al-Gazali LI, Aithala GR, Nork M. Joubert’s syndrome: new cases and review of clinicopathologic correlation. Pediatr Neurol 1999;20:274-281.
19. Boltshauser E and Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie 1977;8:57-66.
20. Steinlin M, Schmid M, Landau K, Boltshauser E. Follow-up in children with Joubert syndrome.Neuropediatrics. 1997;28:204–211. doi: 10.1055/s-2007-973701
21. Desmet VJ: Congenital diseases of intrahepatic bile ducts: variations on the theme "ductal plate malformation". Hepatology 1992, 16:1069-1083.
22. Poretti A, Vitiello G, Hennekam RC, Arrigoni F, Bertini E, Borgatti R et al. Delineation and diagnostic criteria of Oral-Facial-Digital Syndrome type VI. Orphanet J Rare Dis. 2012 Jan 11;7:4. doi: 10.1186/1750-1172-7-4.
23. Doherty D, Glass IA, Siebert JR, Strouse PJ, Parisi MA, Shaw DW, et al. Prenatal diagnosis in pregnancies at risk for Joubert syndrome by ultrasound and MRI. Prenat Diagn. 2005;25:442–47.
|