Introduction
Oral cancer is the sixth most form of common cancer in the world with a 5-year survival rate of less than 50%.[1] The most common causes of oral cancer are tobacco and alcohol, factors that can be controlled. The presentation of oral cancer is typically as an ulcer, red patch or white lesion. It is important for the dental health care provider to inspect the mouth for suspicious lesions and take a biopsy to establish the diagnosis. Since smaller oral cancers have a better prognosis than larger ones, early detection plays an important role in reducing both the treatment associated morbidity and death rate from the disease.[2]
Epithelial carcinogenesis is a multistep process in which an accumulation of genetic events within a single cell line leads to a progressively dysplastic cellular appearance, deregulated cell growth, and, finally, carcinoma.[3] or Oncogenesis (carcinogenesis) is the progression from a normal healthy cell to a pre-malignant or a potentially malignant cell - characterised by an ability to proliferate autonomously. Oncogenesis involves a series of genetic steps and also epigenetic –outside the gene - changes. These changes include the aberrant expression and function of molecules regulating cell signalling, growth, survival, motility, angiogenesis (blood vessel proliferation), and cell cycle control.
The fundamental but grossly oversimplified concept of the genetic mechanism behind cancer is the over-expression of oncogenes and/or the silencing of tumour suppressor genes (TSGs). Cell cycle control is disturbed particularly by over-expression or over-activity (amplification) of oncogenes which drive cell proliferation. Working more towards cell protection are the TSGs. One of the most important of the TSGs is P16 which acts as a checkpoint in growth control. Another important TSG is P53, which will usually either repair a potentially malignant cell or it will destroy it (by apoptosis). However, multiple oncogenes and TSGs are involved in carcinogenesis. Microarray technology has given researchers the ability to produce large amounts of data from nearly the entire known human genome, and has shown that many changes in genes can be involved in oncogenesis. Changes in over one hundred genes have now been implicated in some cancers.[4]
Epidemiology
Oral cancer most commonly occurs in middle-aged and older individuals, although a disturbing number of these malignancies is also being documented in younger adults in recent years. From an epidemiological and clinicopathological perspective, “oral cancer” can be divided into three categories: carcinomas of the oral cavity proper, carcinomas of the lip vermilion, and carcinomas arising in the oropharynx. Intraoral and oropharyngeal tumors are more common among men than women, with a male:female ratio of over 2:1. However, the disparity in the male:female ratio has become less pronounced over the past half century, probably because women have been more equally exposing themselves to known oral carcinogens such as tobacco and alcohol.[5]
Risk Factors
The use of tobacco and alcohol are the two most important risk factors for cancer of the mouth. Smoking is the single most important risk factor for oral cancer. The use of tobacco and alcohol synergistically increase the risk of oral cancer.[1] Although it has been suggested that chewing tobacco is a significant cause of oral cancer. By contrast, the use of pan (areca nut and tobacco leaves soaked in lime) is significantly associated with the development of oral cancer. Although a number of other factors have been implicated in the development of oral cancer, such as various bacterial and viral infections, to date, none have been conclusively proven. For cancer of the lip, the single most important risk factor is heavy exposure to ultraviolet rays of the sun.[2]
Causes Of Oral Cancer
Two separate lines of research are now converging to unravel the complex series of events that lead to cancer. One area has clearly identified site-specific alterations of oncogenes such as EGFR and tumor suppressor genes such as p53. The other is based on epidemiological evidence that has linked exposure to exogenous agents to the development of specific forms of cancer. For example, epidemiological studies have strongly implicated chemical carcinogens, such as those in tobacco, with lung and laryngeal cancer. Exposure to ultraviolet light has been strongly associated with carcinoma of the lower lip. Additionally, evidence is emerging for the role of specific viruses in cancers such as those arising in hematopoietic and lymphoid tissues and those of the uterine cervix.[6] (Fig 1)
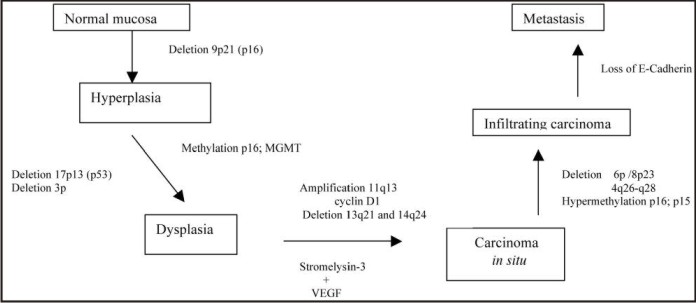 | Fig 1 : Sequence Of Genetic And Tumor/Stroma Alterations Implicated In Origin Of Oral Cancer.7
 |
1. Chemical carcinogens
Animal studies have shown that the application of certain chemicals, such as DMBA, to the oral mucosa will induce the formation of squamous cell carcinoma. However, the link between chemical carcinogens that theoretically might be encountered in daily life such as those ingested in drinking water and oral cancer is not known.[2]
2. Tobacco
Cigarette smoking is well established as an important risk factor in oral cancer. Tobacco smoke contains a large number of chemical carcinogens including aromatic hydrocarbons such as benzopyrene and nitrosamines. These carcinogens have been shown to induce specific genetic changes of the p53 and H-ras genes.[2]
3. Alcohol
All forms of alcohol have been implicated in the development of oral cancer. Importantly, the effects of tobacco and alcohol are additive with alcohol acting synergistically to promote the carcinogenic effects of tobacco products. The mechanism by which alcohol contributes to oral cancer is not well understood but it probably acts directly on the epithelial cells of the oral mucosa by increasing permeability and through its dehydrating effects. In addition, there may also be an indirect effect via altered liver metabolism.4 Interestingly, there is some experimental evidence that alcohol might act to alter the p53 gene directly.[2]
4. Sunlight
Actinic radiation has long been associated with cancer of the lower lip. Ultraviolet light is a potent DNA damaging agent inducing DNA cross-linking, single strand and double strand DNA breaks and nucleotide substitution.[2]
5. Virus infections
• Human papillomavirus infections
Currently more than 100 types of human papilloma viruses (HPV) are known. HPV-6, -11, -16, -18, -31, -33, and -42 have been isolated from the oral cavity. HPV-16 and HPV-18 and others are regarded as carcinogenic. HPV-16 and -18 have been the most common virus types identified in oral carcinoma. HPV-associated oropharyngeal cancers appear to be less associated with tobacco or alcohol use, but more associated with marijuanha and oral sex, and to have a better prognosis.
• Herpes viruses
Herpes simplex viruses (HSV) have also been associated with carcinogenesis. HSV nucleic acids have been found in lip cancer, antibody levels to HSV-1 and -2 are higher in oral cancer patients when compared with controls, and HSV seropositivity together with smoking has been associated with increased cancer risk.
Epstein-Barr virus has also been implicated in oral cancer but the evidence thus far is controversial.[4] (Fig 2).
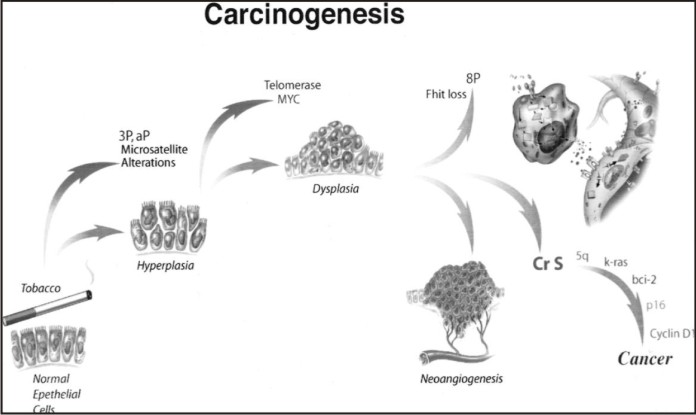 | Fig 2 : Multistep Carcinogenesis Model.3
|
Chromosomal Instability
The most widely used molecular approach to the loss of genetic material is the study of loss of heterozygosity (LOH) based on the detection of differences between normal maternal or paternal alleles in constitutional (normal) DNA and their homonyms in tumour tissue. When a multicellular organism inherits a mutation in an allele of a tumour suppressor gene, it is considered a heterozygote of that gene, i.e. it possesses a mutated (mutated allele) and a normal (normal allele) copy. When a somatic cell with this inherited mutation has a second mutation in the other allele of the suppressor gene, they are both mutated and there is a complete loss of normal protein function, a lack of suppressor function. This is a case of LOH, since both alleles are mutated and the cell or cells that derive from them will therefore be homozygotes (both alleles mutated).[7]
Epigenetic Alterations
There is considerable interest in the role of epigenetic alterations in cancer, explaining their separate consideration in this review. These alterations affect or inactivate gene function by methylation of the promoter region, without changing the structure or sequence of the gene. Methylation is an epigenetic modification by which gene activity is controlled by addition of methyl groups (CH3) to certain DNA cytosines.[8] Most methylations occur in cytosines of CpG nucleotides (cytosine and guanine separated by a phosphate) and are present in a normal manner in promoter regions of certain genes. CpG islands act directly by inhibiting the binding of transcription factors to the DNA or by recruiting proteins that activate histone deacetylases (HDAC), which contain co-repressor complexes additional to DNA methylation. This phenomenon is the main epigenetic modification in humans and methylation pattern changes may play a very important role in carcinogenesis[9] because they are frequently related to the loss of gene expression. They also appear to be essential for the occurrence of the multiple genetic events required for tumour progression, since they can inactivate DNA-repair genes. Both hypermethylation (by suppressor gene inactivation) and hypomethylation (by inappropriate oncogene activation) can produce carcinogenesis.[10]
Tumour - Stroma Interactions
Development by tumour cells of the ability to infiltratesurrounding tissue and produce metastasis is one of the finalsteps in tumour progression. This ability depends on destructionof the extracellular matrix (ECM), other stromal componentsand the basement membrane. Matrix metalloproteinases (MMPs) form a family of zinc dependent endopeptidases thatappear to play a major role in ECM degradation and in tumourinvasion and metastasis.[11] MMP-2, MMP-9 and MMP-3have all been found to be related to tumour invasion, a moreaggressive growth and a worse prognosis. expression of these genes could be used as tumour progression marker.[12]
Oncogenes
The over-expression of oncogenes -such as the epidermal growth factor receptor (EGFR) gene – can promote growth, survival, and spread of cells - leading to the development of cancer. The range of oncogenes identified is quite broad, and the mechanisms by which they act are complex.[4]
Numerous oncogenes have been implicated in oral carcinogenesis. Aberrant expression of proto-oncogene epidermal growth factor receptor, (EGFR/c-erb 1) and members of gene families ras, c-myc, int-2/Fgf-3 (fibroblast growth factor-3), hst 1/HSTF1 (heparin-binding secretory transforming factor 1), PRAD-1 (parathyroid adenomatosis 1), and bcl-1(B-cell leukemia/lymphoma 1) is believed to contribute to cancer development.[12]
Tumour Suppressor Genes
Although oncogenes alone are not able to cause oral cancer, they appear to be initiators of the process. The crucial event in the transformation from premalignancy to malignancy is inactivation of negative cell regulators (tumour suppressor genes). Tumour suppressor genes are frequently inactivated by point mutations, deletions and rearrangements in both copies of the gene.[12],[13] Among the most widely studied tumour suppressor genes are rb and p53, which express pRb (retinoblastoma protein) and p53 protein respectively. These proteins control the cell cycle and are involved in the inhibition of cell proliferation.[7] TSG function can be disturbed by aberrations such as deletions or mutations in TSG genes, or by TSG silencing from hypermethylation, and any of these changes can lead to cancer.[4]
Single Nucleotide Polymorphisms (Snps)
Single nucleotide polymorphisms (SNPs) are gene areas that have altered DNA sequences which may not lead to an aminoacid alteration, or altered DNA sequences that do not seem to have any adverse effect in ‘normal’ individuals but may be markers for disease predisposition, or may be used to genetically identify patients, as they tend to cluster with ethnic background. SNPs in TSGs, xenometabolising enzymes, and DNA repair enzymes may also play a role in cancer development - at least in some individuals.[4]
Early Diagnosis
There is abundant evidence that early diagnosis would reduce the morbidity and mortality from oral cancer. Increased clinical suspicion and the introduction of diagnostic aids may help achieve earlier diagnosis. The drawbacks of conventional histopathology are becoming evident, along with the human limitations in diagnosis in this as in many other fields of medicine. This is especially the case in tissue biopsies from close to previous suspicious lesions in the upper aerodigestive tract.[12] Hopefully, molecular examination of tissue might prove useful in, for example, examining biopsies from PMDs; from tumour surgical margins that appear histologically clear, and to detect clinically undetected lymph node metastases.[4] The most predictive of the tissue molecular markers thus far available and assessed in oral cancer development[15] include the TSG p53 protein expression, and changes in chromosomes 3p or 9p (probably due to changes in the TSG p16).
The use of these markers as an adjunct to routine histopathological examination may eventually help in prognostication and effective management of these lesions but routine use is hampered thus far by the cost and complexity of the tests, the lack of facilities in some laboratories, and limited outcome studies.[16]
Prevention
At least three-quarters of oral cancers could be preven-ted by the elimination of risky lifestyles such as tobacco smoking and alcohol consumption. Smoking cessation contributes to reducing the risk of oral cancers, with a 35% reduction in risk within 1-4 years and 80% reduction of risk by 20 years, reaching the level of never smokers.Treatment of tobacco dependence is an important step to reduce oral cancer in high risk groups. Tobacco cessation among high-risk patients ie those with potentially malignant disorders need to be addressed through the primary care practitioners (including dentists) and where possible with assistance from specialist smoking cessation clinics. Protection against solar irradiation could further reduce the incidence of lip cancers.[4]
References
1. Napier SS, Speight PM. Natural history of potentially malignant oral lesions and conditions: an overview of the literature. J Oral Pathol Med.(2008) ;37(1):1-10.
2. Jordan RC. Oral Cancer. Crest® Oral-B® at dentalcare.com Continuing Education Course, November 1(2009).
3. Tsao AS, Kim ES, Hong WK. Chemoprevention of Cancer. CA Cancer J Clin (2004) ;54: 150–180.
4. Scully C. Oral cancer aetiopathogenesis; past, present and future aspects.Med Oral Patol Oral Cir Bucal. (2011); 16(3): 306-311.
5. Neville BW. Oral Cancer and PrecancerousLesions. CA Cancer J Clin (2002) ;52: 195-215.
6. Lingen MW, Kalmar JR, Karrison T, Speight PM. Critical evaluation of diagnostic aids for the detection of oral cancer. Oral Oncol. (2008); 44(1):10-22.
7. J. Campo-Trapero, J. Cano-Sanchez , B. Palacios-Sanchez, JJ. Sanchez-Gutierrez, M.A. Gonzalez-Moles and A. Bascones-Martinez. Update on Molecular Pathology in Oral Cancer and Precancer. Anticancer Research (2008); 28: 1197-1206.
8. Shaw R: The epigenetics of oral cancer. Int J Oral Maxillofac Surg (2006); 35: 101-108.
9. Rosas SL, Koch W, Costa Carvalho MG, Wu L, Califano J, Westra W, Jen J and Sidransky D: Promoter hypermethylation patterns of p16, O6-methylguanine-DNA-methyltransferase, and death-associated protein kinase in tumors and saliva of head and neck cancer patients. Cancer Res. (2001); 61: 939-942.
10. Auerkari EI: Methylation of tumor suppressor genes p16(INK4a), p27(Kip1) and E-cadherin in carcinogenesis. Oral Oncol (2006)42: 5-13.
11. Kim MM and Califano JA: Molecular pathology of head-and neck cancer. Int J Cancer (2004);112: 545-553.
12. Williams HK: Molecular pathogenesis of oral squamous carcinoma. Mol Pathol.(2000); 53: 165-172.
13. Bascones-Ilundain C, Gonzalez-Moles M, Campo-Trapero J, Gil-Montoya J, Esparza-Gomez G, Cano-Sanchez J and Bascones-Martinez A: No differences in caspase-3 and Bax expression in atrophic-erosive vs. reticular oral lichen planus. J Eur Acad Dermatol Venereol (2008); 22: 204-212.
14. Fischer DJ, Epstein JB, Morton TH Jr, Schwartz SM. Reliability of histologic diagnosis of clinically normal intraoral tissue adjacent to clinically suspicious lesions in former upper aerodigestive tract cancer patients. Oral Oncol. 2005;41:489-96.
15. Lee JJ, Hong WK, Hittelman WN, Mao L, Lotan R, Shin DM, et al. Predicting cancer development in oral leukoplakia: ten years of translational research. Clin Cancer Res. (2000) ;6:1702-10.
16. Scully C, Sudbø J, Speight PM. Progress in determining the malignant potential of oral lesions. JJ Oral Pathol Med. (2003) ;32: 251-256.
|