Introduction
The Sturge-Weber Syndrome (SWS) or encephalotrigeminal angiomatosis, is specifically congenital, non-hereditary condition of rare development. It is the most frequent neurocutaneous syndrome, with the proportion of 1/50,000 births[1],[2] It belongs to a group of disorders collectively known as the phakomatoses (‘‘mother spot disease’’). The predominant clinical manifestations of SWS include cutaneous manifestations like unilateral cutaneous vascular nevus (following the V1 and V2 division of the 5th cranial nerve) and neurological manifestations including progressive seizures, ipsilateral glaucoma, hemiatrophy, contralateral hemiparesis, hemianopia and mental retardation. The syndrome results from malformation of the cerebral vasculature located within the pia mater, most commonly over the occipital region[3]. These malformations lead to venous hypertension and subsequent hypoperfusion of the underlying cortex, causing chronic cerebral ischemia, atrophy and neurological deterioration[4].
Oral changes are noted in 40% of cases in the form of angiomatosis of lips causing macrochelia and hemiatropohy of buccal mucosa, floor of mouth and palatal mucosa. Other abnormalities reported are severe gingival enlargement, pyogenic granulomas, unilateral alveolar hypertrophy, ipsilateral premature or delayed eruption and malocclusion. Because of neurological changes, many patients with SWS find it difficult to maintain adequate oral hygiene, leading to an increased risk of periodontal disease and caries. In addition, the medication usually employed to manage their seizures may lead to gingival hyperplasia.
Case Report
A 14 year old male child reported to the dental clinic with the chief complaint of pain and swelling in upper lip since last 2 - 3 months. His past history revealed diffuse reddish bluish stain present on the left side of face since birth. Redness was present since 4 years in the left eye with partial loss of vision. Swelling and mild pain was present in left upper lip since 3 years. Swelling of lip had neither increased nor decreased in size since then. Headache was present since 2 years which was severe, and intermittent in nature. No history of epilepsy, mental retardation, or paraesthesia’s was present.
Extra oral examination
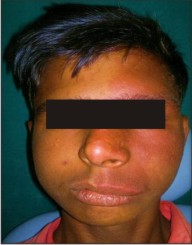
Facial asymmetry of mid face was present due to swelling and drooping of upper lip (Fig- 1). Unilateral diffuse reddish blue macular stain present on left side of upper and middle third of the face extending from midline to 2-3 cm in front of left ear anteroposteriorly and from forehead to corner of left side of mouth superioinferiorly (Fig - 1 & 2).
 | Figure 1
 |
 | Figure 2
|
The discoloured patch was irregular in shape, and the margins of the patch were well defined with smooth surface. Swelling was present in upper left vermillion border of the lip which was red in colour. Redness of the eye was there on the affected side. On Palpation, the patch was not raised and was smoothly merging with the normal skin. The surface was smooth, did not blanch on pressure, and no tenderness was elicited on applying pressure. The patch was warm to touch. No pulsations were felt.
Intra Oral Examination
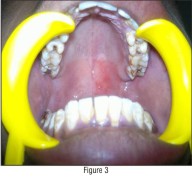
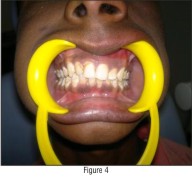
A unilateral diffuse reddish discolouration was present on left mucosa and soft palate with a distinct line separating the two halves (Fig - 3 & 4).
 | Figure 3
|
 | Figure 4
|
Based on the clinical findings a provisional diagnosis of vascular angioma was given with differential diagnosis as arteriovenous malformation ( capillary ) and sturge weber syndrome.
Radiographic Assessment:
Lateral skull radiograph revealed no pathology. CT skull revealed unilateral calcifications in the left occipital region (Fig - 5 & 6).
 | Figure 5
|
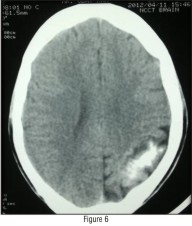 | Figure 6
|
Patient was sent for ophthalmologist opinion and report suggested of ciliary congestion, temporal pallor, cupping of fundus with shrinking of blood vessels of left eye indicative of glaucoma.
A final diagnosis of STURGE WEBER SYNDROME (Type I) was given based on his clinical findings and radiological examination.
Discussion:
Schirmer described the first report of the syndrome in 1860 in a patient with bilateral facial nevus and unilateral buphthalmos. In 1879, William Allen Sturge presented another case of a 6-year-old girl with bilateral facial nevus, congenital glaucoma and vascular malformation at the right eye and progressive twitching on the left side of her body. In 1922, Parkes Weber was the first to demonstrate the associated intracranial calcifications in this syndrome radiographically[5]. The cutaneous angioma is called a port-wine stain (PWS). The primary complications involving the ipsilateral eye are buphthalmos and glaucoma. The neurologic manifestations may vary, depending on the location of the LAs, which most commonly are located in the parietal and occipital regions and the secondary effects of the angioma. Neurological manifestations may include
1) Seizures, which may be intractable; focal deficits, such as hemiparesis and hemianopsia, both of which may be transient, called "strokelike episodes"
2) Headaches; and
3) Developmental disorders, including developmental delay, learning disorders, and mental retardation[6].
SWS is referred to as complete when both CNS and facial angiomas are present and incomplete when only 1 area is affected without the other. The Roach Scale[7] is used for classification, as follows:
Type I - Both facial and leptomeningeal angiomas; may have glaucoma
Type II - Facial angioma alone (no CNS involvement); may have glaucoma
Type III - Isolated LA; usually no glaucoma
In our case the patient had facial port wine stain with history of headache (learning disability was present with no history of epilepsy) and glaucoma, hence he was classified as type 1 on Roach scale.
Pathophysiology :
SWS is caused by residual embryonal blood vessels and their secondary effects on surrounding brain tissue. A vascular plexus develops around the cephalic portion of the neural tube, under ectoderm destined to become facial skin. Normally, this vascular plexus is formed in the sixth week and regresses around the ninth week of gestation. Failure of this normal regression results in residual vascular tissue, which forms the angiomata of the leptomeninges, face, and ipsilateral eye. Neurologic dysfunction results from secondary effects on surrounding brain tissue, which include hypoxia, ischemia, venous occlusion, thrombosis, infarction, or vasomotor phenomenon[8]. A “vascular steal phenomenon” may develop around the angioma, resulting in cortical ischemia. Therefore, recurrent seizures, status epilepticus, intractable seizures, and recurrent vascular events may aggravate this steal further, with an increase in cortical ischemia, resulting in progressive calcification, gliosis, and atrophy, which in turn increase the chance of seizures and neurologic deterioration[9]. Disease progression and neurologic deterioration may occur in SWS. Although the actual LA is typically a static anatomic lesion, some authors have clearly documented the progressive nature of SWS[10]. The main ocular manifestations (ie, buphthalmos, glaucoma) occur secondary to increased intra ocular pressure with mechanical obstruction of the angle of the eye, elevated episcleral venous pressure, or increased secretion of aqueous fluid.
Diagnosis:
The diagnosis is based on the presence of the nevus flammeus in the face, and, according to some authors, the association between convulsion and the nevus, irrespectively of the neurological loss, is sufficient for the syndrome's clinical diagnosis. Ophthalmologic changes may be observed through the retinal examination and also ocular ultrasonography. CSF analysis may reveal elevated protein due to micro haemorrhages. Skull films may reveal tram track calcification caused by calcification in opposing gyri, ipsilateral calvarial thickening and enlargement of the paranasal sinuses and mastoid. Cranial CT demonstrates cortical atrophy underlying the angioma with gyriform “tram track” calcification. MRI is the current “gold standard” for diagnosis of disease which is reliable even in very young infants. as the cerebral vascular malformations are more easily viewed through Magnetic Resonance imaging. Functional cerebral studies using PET and SPECT have shown abnormalities of metabolism and perfusion in Sturge Weber syndrome.
Differential Diagnosis
Capillary Malformations These are typically referred to as Port Wine Stains and can be classified according to the Waner Grading system. Grade I lesions with vessel diameters are in the 80 µgm range. These lesions are light pink macules. Grade II lesions with vessel diameters measure up to 120 µgm. these lesions are darker pink macules. Grade III lesions are vessel diameters measure up to 150 µgm. These lesions are red macules. Grade IV lesions are vessel diameters greater than 150 µgm. These lesions are purple and may become papular.
Klippel-Trenaunay-Weber Syndrome includes capillary haemangiomas of the extremities and face, congenital venous abnormalities, hemihypertrophy of soft and bony tissues, in addition to all the characteristics of SWS.
Rendu-Osler-Weber's Syndrome or hereditary hemorrhage telangiectasis is an abnormal vascular dilatation of the skin terminal vessels, mucosa and viscera. The disease involves the lips, buccal mucosa, pharynx, conjunctives and face, in addition to neuro fibromatosis, elliptocytosis and arteriovenous aneurysm.
Mortality and Morbidity
Neurologic and developmental morbidity includes seizures, weakness, strokes, headaches, hemianopia, mental retardation, and developmental abnormalities. The development of seizures and the age of onset may correlate with the degree of neurologic involvement. Neurologic dysfunction increases with bilateral PWS. Patients may experience complications related to refractory seizures and anticonvulsants, visual loss and blindness from glaucoma, cosmetic deformities, and other manifestations of soft-tissue involvement. Of 60 patients in the combined series from Children's Hospital, Boston, 2 deaths (3.3%) have been reported. Erba and Cavazutti, reported occurrence of 1 death in the postoperative period, after epilepsy surgery; in another study, 1 death occurred secondary to intractable seizures. Oakes reported 4 deaths in 30 patients[11].
Management
This includes anticonvulsants for seizure control, symptomatic and prophylactic therapy for headache, glaucoma treatment to reduce the intra ocular pressure, and laser therapy for PWS.
Seizures: Since the seizures are typically focal, an anticonvulsant with efficacy in focal seizures is preferable.
Glaucoma: The goal of treatment is control of IOP to prevent optic nerve injury. Medications either decrease the production of aqueous fluid or promote the outflow of aqueous fluid. Beta-antagonist eye drops reduce the production of aqueous fluid, Adrenergic eye drops and miotic eye drops reduce IOP by promoting drainage, and Carbonic anhydrase inhibitors decrease IOP by decreasing production of aqueous fluid.
Headaches: Recurrent headaches can be treated with symptomatic and prophylactic medications. Various agents used are acetaminophen, ibuprofen, gabapentin, sodium valproate, amitriptyline. Aspirin also has been used for headaches and to prevent vascular disease.
Port Wine Stain: These need to be evaluated within the first week of life and differentiated from hemangioma. PWS are treated with laser therapy, which should start as soon as possible, since multiple treatments are needed and earlier treatment may reduce the number needed. Also, the smaller the lesion initially, the fewer the laser flashes needed to remove the lesion. Surgery is desirable for refractory seizures, glaucoma, and specific problems related to various associated disorders, such as scolios.
Conclusion
This syndrome is not much frequent, but it needs to be early diagnosed, since it brings a series of complications to its carriers when not treated, especially because of reaching the Central Nervous System. The health professionals have to be suitably able to recognize its characteristic signs and symptoms, and so improve the quality of life of these patients.
References
1. Gomes ACA, Silva EDO, Albert DGM. Síndrome de Sturge-Weber: Relato de caso clínico. Revista de Cirurgia e Traumatologia Buco-Maxilo-Facial. 2004 4: 01-71.
2. Cronemberger S, Calixto N, Assunção DAM, Milhomens EG. Síndrome de Sturge-Weber: estudo ultrabiomicroscópico. Arquivo Brasileiro de Oftalmologia. 2004, 67:211-217.
3. Sadda SR, Miller NR, Tamargo R, Wityk R. Bilateral optic neuropathy associated with diffuse cerebral angiomatosis in Sturge–Weber syndrome. J Neuroophthalmol 2000;20(1): 28-31.
4. Paller AS. The Sturge–Weber syndrome. Pediatr Dermatol 1987;4(4): 300-4.
5. Pearce JM. Sturge Weber Syndrome (encephalotrigeminal or leptomeningeal angiomatosis). J Neurol Neurosurg Psychiatry 2006; 77: 1291-1292
6. Bodensteiner JB, Roach ES. Sturge-Weber Syndrome: Introduction and Overview. In: Bodensteiner JB, Roach ES, eds. Sturge-Weber Syndrome. Sturge Weber Foundation. Mt Freedom, New Jersey. 1999.
7. Roach ES. Neurocutaneous syndromes. Pediatr Clin North Am. Aug 1992; 39(4):591-620.
8. Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. Aug 2003;18(8):509-16.
9. Okudaira Y, Arai H, Sato K. Hemodynamic compromise as a factor in clinical progression of Sturge- Weber syndrome. Childs Nerv Syst. Apr 1997;13(4):214-9
10. Maria BL, Neufeld JA, Rosainz LC. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. J Child Neurol. Dec 1998;13(12):606-18.
11. Oakes WJ. The natural history of patients with the Sturge-Weber syndrome. Pediatr Neurosurg. 1992;18(5-6):287-90
|