Introduction
The Ectodermal Dysplasia’s compromise a large, heterogeneous group of inherited disorders that are defined by primary defects in the development of two or more tissues derived from embryonic ectoderm. The tissues primarily involved are the skin, hair, nails, eccrine glands and teeth. It was first described by Thurman in 1848 and the term Ectodermal dysplasia was coined in 1929 by Weech.To date, more than 192 distinct disorders have been described .The most common ED are X-linked recessive hypohidrotic ectodermal dysplasia (Christ- Siemens-Touraine syndrome) and hidrotic ED (Clouston syndrome).
The first classification of ED was proposed by Freire-Maia and Pinheiro in 1982[4] with additonal updates in 1994 and 2001[5],[6]. Originally classification stratified the ED into different subgroups according to the presence or absence of
-
Hair abnormalities or trichidysplasia
-
Dental abnormalities
-
Nail abnormalities or onychodysplasias
-
Eccrine gland dysfunction or dyshidrosis.
Overall the ED were classified into either group A disorders, which were manifested by defects in at least 2 of the 4 classic ectodermal structures and group B disorders which were manifested by a defect in one classic ectodermal structure (1-4 from above) in combination with (5) a defect in one other ectodermal structures (i.e. ears, lips dermatoglyphics). Eleven group A subgroups were defined, each with a distinct combination of 2 or more ectodermal defects (e.g. 2-4, 1-2-3, from above). The group B disorders were indicated as 1-5, 2-5, 3-4, 4-5 from above. In 2003 Lamartine[7] reclassified ED into four functional groups based on the underlying pathophysiologic defect:
1. Cell to cell communication and signaling
2. Adhesion
3. Development
4. Other
Similarly in 2001, Priolo and Lagana [8] reclassified the E.D into 2 main functional groups:
1. Defects in developmental regulation / epithelial mesenchymal interaction and
2. Defects in cytoskeleton maintenance and cell stability,
Several ED may manifest in association with midfacial defects, mainly cleft lip, cleft palate or both. The 3 most commonly recognized entities are:
1. ED, ectrodactyly or clefting (EEC syndrome).
2. Hay Wells Syndrome or ankyloblepharon,ED and cleft lip / palate (AEC syndrome) and
3. Rapp-Hodgkin Syndrome, all of which are caused by mutations in the TP63 gene.
Although more useful is the clinically relevant classification that divides Hereditary ED into 2 broad cateogries[8].
1. X-Linked Hypohidrotic form (Christ- Siemens Touraine Syndrome),characterized by classic triad of hypodontia, hypohidrosi,hypotrichosis and by characteristic dysmorphic facial features:
a. The typical faecies, which is often not recognized until infancy, is characterized by frontal bossing, sunken cheeks, saddle nose, thick everted lips, wrinkled hyper pigmented per orbital skin and large low set ears.
b. Dental manifestation include conical or pegged teeth, hypodontia or complete anodontia and delayed eruption of permanent teeth.
c. Most patients have fine sparse lusterless, fair hair, therefore either pigmentation in the hair shaft is observed microscopically and the medulla is often discontinuous. When medullation is present “bar code “appearance is seen.
d. Onychodystrophy may occur but is not common.
e. Extrinsic scaling of the skin and unexplained pyrexia secondary to anhidrosis may occur in the neonate period. The development of a chronic eczematous dermatitis is common.
f. Other common signs are short stature, eye abnormalities, decreased tearing and photophobia.
2. Hidrotic form (Clouston Syndrome): it usually spares the sweat glands but affects the teeth, hair and nails .Most of other clinical features are similar to that seen in the hypohidrotic form. It has an AD inheritance and is common in persons of French-Canadian ancestry.
Christ-Siemens-Touraine Syndrome with X-linked recessive inheritance is the most frequently reported manifestation of ED.Depending on the severity of clinical manifestation; Christ-Siemens-Touraine syndrome can be classified as either hypohidrotic or anhidrotic ED1.
We present a case of X- linked recessive hypohidrotic ED.
Case Report
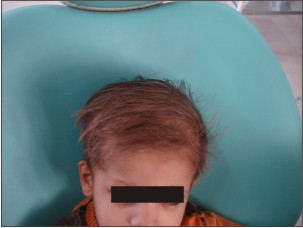
Parents of 3 yr old male patient reported to the Department of Oral Medicine and Radiology, Inderprastha Dental College with a chief complaint of missing teeth. On further questioning, parents revealed that only two front and back teeth were present, that caused difficulty in chewing foofd.Also the shape of upper front teeth was abnormal. There was no past history of trauma and dental extractions. Parents did not report of any diseases or drugs taken; although the parents complained of frequent history of pyrexia and inability to bear heat. The condition aggravated in the hot summer months and the patient had to be kept in an air-conditioned room. Patient was y youngest of the 3 siblings, other siblings were females aged 11 and 7yrs. None of the siblings reported any problems. There was no previous family history in either of the parents. The child had normal intelligence and no visual and auditory problems. On extra oral examination ,patient had a typical faecies, frontal bossing, saddle nose,protruberant lips (Fig 1),thinning and light colored scalp hair(Fig 2), absence of hair on arms and legs (Fig 3).On intraoral examination ,peg shaped maxillary deciduous central incisors were present(Fig 4). Maxillary second deciduous molars were the only teeth present,mandibular ridge was edentulous(Fig 5); loss of vertical bone height was noticed. Panoramic radiograph was taken that revealed the absence of deciduous and permanent teeth (Fig 6). Based on the history, clinical examination and radiological examination we arrived at a diagnosis of X-linked hypohidrotic Ectodermal Dysplasia. Was advised a prosthetic rehabilitation to improve appearance and function.
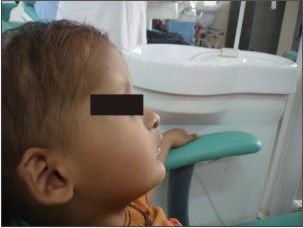 | Figure 1 : Shows Profile View Of The Patient.
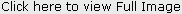 |
 | Figure 2 : Shows Thinning Of Scalp Hair.
|
 | Figure 3 : Shows Absence Of Hair On Hands And Feet.
|
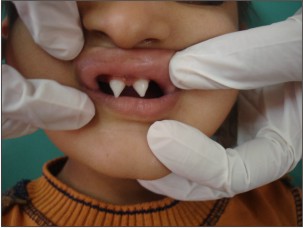 | Figure 4 : Shows Peg Shaped Maxillary Central Incisors.
|
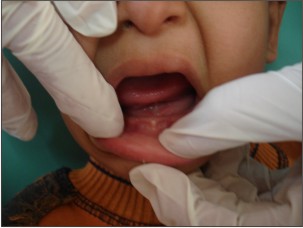 | Figure 5 : Shows Edentulous Mandibular Ridge.
|
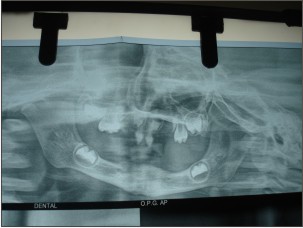 | Figure 6 : Shows Opg With Missing Teeth.
|
Discussion
The genetic defects responsible for approximately 30 of ED have been identified. Genetic studies regarding the etiology of ED reveal that mutation in the ectodysplasin A and ectodysplasin A receptor genes are responsible for X- linked and autosomal hypohidrotic ED1.Ectodysplain is important in promoting cell survival,growth and differentiation.AD and AR Hypohidrotic ED are caused by mutation in the DL gene , which encodes EDA receptor ( ectodysplasin).AR hypohidrotic ED may result from mutations in the EDARADD gene, which encodes a protein that interacts with EDA receptor.
Hidrotic ED(Clouston syndrome) which is an AD disorder is caused by mutation in GJB6 which encodes connexin 30 , a component of intercellular gap junction.X-linked recessive hypohidrotic ED has full expression only in males.Female carrier outnumber affected men ,but females show little or no signs of these condition; as in our case. The diagnosis of ED is made when at least 2 types of abnormal ectodermal features occur such as malformed teeth and extremely sparse hair [2],[3]. like in present case. Lab studies are useful in the diagnosis or management of ED .Although to evaluate dental abnormalities, OPG can be done at an early age. Sweat pore counts using yellow starch iodine, pilocarpine iontophoresis and skin biopsy may document hypohidrosis and a reduction in the number of eccrine gland s.Genetic testing is available through GeneDx.
Differential diagnosis includes alopecia areata, aplasia cutis congenital, focal dermal hypoplasiasyndrome, incontinentia pigmenti, Naegli Frranceschetti Jadassohn syndrome and Pachyonchia Congenita.
At present, no pharmacological treatment is available for ED.However patients with hypohidrosis/anhydrisois; airconditioning for home, school and work is advised. Avoid vigorous physical activities .Antipyretics are not effective in treatment of hyperpyrexia. Similar was the case with our patient who had frequent bouts of hyperpyrexia and heat tolerance.’ Patients affected by anodontia may show shrinkage of bone supporting the denture after long term denture use. The prognosis for most patients with ED is very good. Oral rehabilitation is necessary to improve both the saggital and vertical skeletal relationship during craniofacial growth and development as well as to provide improvements in esthetics, speech and masticatory effeciency[9].
Conclusion
The clinical manifestations of ED cause considerable social problem in individual affected by the condition. Treatment should be administered by a multidisciplinary team involving pediatric dentistry, orthodontics, prosthodontics and oral surgery.
References
1. Mehmat Bani, Ali Mein Tezkirecioglu, Nese Akal.Ectodermal dysplasia with anodontia: a report t of 2 cases: Eur.J.Dent 2010 April; 4(2):215-222.
2. Vieira KA,Teixerra MS,Guirado CG,Gravio MB-P rosthodontic treatment of hypohidrotic ectodermal dysplasia with complete anodontia-a case report.Quintessence Int 2007;38:75-80.
3. Yavuz KA,Ulku SZ,Kama JD,Kaya S et al-Ectodermal dysplasia :clinical diagnosis.Int Dent Med Disorders 2008;1:1-10.
4. Pinheiro M, Freire Maia N.The ectodermal dysplasia.Arch Dermatol 1982; 118:215-6.
5. Pinheiro M, Freire Maia N.ED: a clinical classification and a causal review.Am.J.Med Gennet 1994; 53:153-62.
6. Freire Maia N, Lisboa Costa T, Pagnan NA.Ectodermal Dysplasia –how many? Am J Med Genet 2001; 104:84.
7. Lamartine J.Towards a new classification of ED.Clin Exp Dermatol 2003; 28:351-5.
8. Deshpande SN, Kumar Vikas.Ectodermal dysplsia –Maxillary and mandibular alveolar reconstruction with dental rehabilitation –a case report and review of literature. Indian .J. Plast.Surg 2010 Jan-Jun; 43(1):92-96.
9. Tarjan I, Gabris K, Rozsa N.Early prosthetic treatment of patients with ectodermal dysplasia: a clinical report .J. Prosthet Dent 2005; 93:419-424.
|