Cardiovascular disease, and in particular atherothrombotic disease causing coronary heart disease (CHD), stroke, or peripheral vascular disease remains the leading cause of death and of severe disability in developed countries.
Atherosclerosis has been defined as a progressive disease process that involves the large to medium-sized muscular and large elastic arteries. The advanced lesion is the atheroma, which consists of elevated focal intimal plaques with a necrotic central core containing lysed cells, cholesterol ester crystals, lipid-laden foam cells, and surface plasma proteins including fibrin and fibrinogen. This central core is associated with a cellular infiltrate with hypertrophic smooth muscle cells, macrophages, and sparse T-lymphocytes. The presence of atheroma tends to make the patient thrombosis prone because the associated surface serves to enhance platelet aggregation and thrombus formation.1
Evidence points to an infectious etiology of cardiovascular disease (CVD), suggesting bacterial or viral infections as potential risk factors for the manifestation of endothelial dysfunction and, in consequence, CVD manifestation.2
Pathogenesis Of Atherosclerosis
Ross3 first highlighted the histological similarities between atherosclerotic plaques and healing inflammatory lesions and proposed a “response to injury” hypothesis for their formation. He proposed that the initial lesion results from injury to the endothelium and leads to a chronic inflammatory process in the artery. This results in the migration of monocytes through the endothelium into the underlying tissue and the proliferation of smooth muscle cells. Activation of the monocytes in the blood vessels leads to the release of hydrolytic enzymes, cytokines, chemokines and growth factors, which induces further damage, leading to focal necrosis. Macrophages also accumulate lipids, especially low density lipoproteins (LDL) in the oxidized or modified form. When the LDL particles are trapped in the artery, they can undergo progressive oxidation and can be internalized by macrophages, with formation of lipid peroxidases and accumulation of cholesterol esters. This results in the production of foam cells. Modified LDL is chemotactic for other monocytes and can induce the production of factors from macrophages that expand the inflammatory response.
A fatty streak can become a fibrous plaque, which becomes complex with a lipid core, calcification, and deposition of extracellular matrix protein. Activated T cells may stimulate metalloproteinase production by macrophages, which remodel the fibrotic plaque and forms a dense fibrous cap. Through remodeling of the extracellular matrix, the fibrous cap may become thin and rupture, leading to activation of the clotting system with thrombosis.4
Mechanisms By Which Periodontal Infections May Contribute To Atherosclerosis
Several possible mechanisms may operate independently or in concert to explain the association between infections in general, and periodontal infections specifically, and atherosclerosis, myocardial infarction, and stroke. These are:
1) Direct effects of infectious agents in atheroma formation
2) Indirect or host-mediated effects triggered by infection
3) Common genetic predisposition for periodontal disease and atherosclerosis
4) Common risk factors, such as lifestyle.
Chun et al5 described various common risk factors implicated in atherogenesis and periodontal disease. These may be non modifiable (gender, age, race, family history, hypertension, diabetes mellitus, body mass index, homocysteine, lipoprotein a, white blood cells, fibrinogen, total cholesterol, low density lipoprotein) and modifiable (lack of exercise, stress, alcohol consumption, diet, smoking, infections, CRP). They further stated that in order to consider periodontal disease as a risk factor for atherosclerosis, the presence of pathogens associated with periodontal disease should be localized in serum or atheromatous plaques.
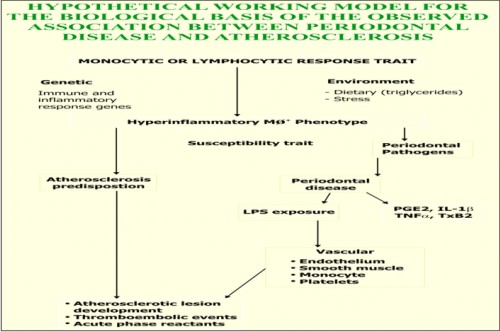
Beck et al1 proposed a hypothetical working model for the biological basis of the association between periodontal disease and atherosclerosis, coronary artery disease and stroke. They said that patients with certain forms of periodontitis possess an MØ+ phenotype. These include early onset periodontitis, refractory periodontitis, insulin dependent diabetes mellitus and atherosclerosis. This phenotype has the following properties:
1) It is a systemic hyperinflammatory monocyte phenotype which secretes abnormally high levels of inflammatory cytokines.
2) It appears to be under both genetic and environmental influences. Monocytic hyper-responsiveness to LPS has been mapped to the area of HLA-DR3/4 or –DQ. Dietary induced elevation of serum LDL has been shown to upregulate monocytic responses to LPS, thereby providing a behavioural or environmental influence on the MØ+ phenotype.
 |
 |
 |
|
Thus periodontal disease, once established, results in a chronic, systemic vascular challenge with bacterial LPS and host-derived inflammatory cytokines that are theoretically capable of initiating and promoting vasculitis and atheroma formation.
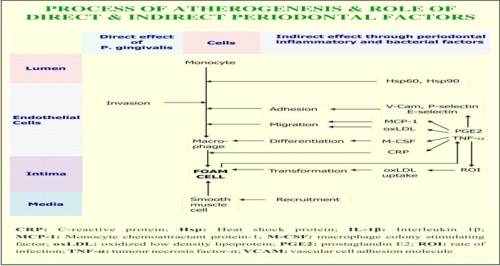
Among periodontal pathogens, Porphyromonas gingivalis has been recognized as a key pathogen and risk factor for periodontal disease. P. gingivalis affects the vessel wall directly and/or indirectly via inflammatory response, immune responses, and haemostasis. The inflammatory host response does not have to be exclusively triggered by P. gingivalis. A current hypothesis suggests an additive or synergistic effect resulting from coincidental bystanders of different origins. Together as total pathogenic burden they might overcome a certain threshold resulting in unequivocal clinical significance. The invasion of P.gingivalis is mediated through upregulation of adhesion molecules, such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), P & E selectins, only in the presence of fimbriae. The activation of adhesion molecules is also required to bind leucocytes to endothelium, which initiates transmigration & atherogenesis.6 Endothelial cell damage has been shown to be promoted by the ability of P. gingivalis to adhere, invade & proliferate in coronary endothelial cells. This interferes with the physiologic dilatory function of of the vessels through the pathogen’s damage of the endothelial & smooth muscle cells. The increased release of proinflammatory cytokines (prostaglandin E2, interleukin 1β and tumour necrosis factor α) actively recruits more inflammatory cells, which furthermore may result in higher counts of foam cells. In addition, the ability of P. gingivalis to stimulate superoxide generation from macrophages and endothelial cells leading to enhanced oxidized LDL generation may augment the localized vascular inflammatory response.7
Kuramitsu et al7 stated that P.gingivalis exhibits several properties which could play a role in CVD as mediators of LDL oxidation, foam cell formation, and rupture of atherosclerotic plaque. They demonstrated that P. gingivalis, as well as its outer membrane vesicles are able to induce foam cell formation in the murine macrophage cell line J774 A.1. This property appears to be mediated by the lipopolysachharide fraction of the cells. Since the rupture of fibrous cap of plaque appears to be an important factor in acute coronary syndrome, it was demonstrated that P.gingivalis 381 degraded fibrous caps isolated from autopsy samples. In addition, it was observed that strain 381 strongly induced matrix metalloproteinase (MMP)-9 protease activity, implicated in plaque rupture from the J774 A.1 macrophages. Finally strain 381 was able to enhance monocyte chemoattractant protein-1 (MCP-1) and NADPH oxidase expression from endothelial cells.
Sharma et al8 reported that P. gingivalis was able to induce platelet aggregation, and that oral strains of Actinobaillus actinomycetemcomitans, Bacteroides forsythus, Campylobacter rectus, Fusobacterium nucleatum, Prevotella intermedia and Trepenoma denticola failed to aggregate platelets under similar conditions. Additionally, they showed that vesicles (outer membrane evaginations that are shed into the environment by the bacteria) of P. gingivalis are potent inducers of mouse platelet aggregation in vitro. They showed that initial adherence of the bacterium to platelet may be facilitated by P. gingivalis fimbriae and P. gingivalis vesicles possess platelet aggregation–inducing activity.
Mattila et al9 discussed the fact that bacterial infections and compounds have profound effect on endothelial cells, monocyte-macrophages, thrombocytes, blood coagulation, and lipid metabolism. They concluded that dental infections are the only factor outside the scope of the classic coronary risk factors that has shown an independent association with the severity of adult coronary atherosclerosis.
According to Renvert et al10 a combination of an anti-infective and mechanical initial therapy with CRx-102 may be beneficial for patients with chronic adult periodontitis. Such a treatment model may even reduce the risk for systemic diseases, such as coronary heart disease, in periodontitis patients. CRx-102 is an oral synergistic combination drug, combining dipyridamole (200 to 400 mg) and low dose prednisolone (3mg). Neither of these compounds is used routinely in periodontal therapy. CRx-102 suppresses the transcription of proinflammatory genes, including cytokines, through a combination of actions that converge at the level of the promoters of these genes. Vidal et al11 concluded that non-surgical periodontal therapy was effective in improving periodontal clinical data and in reducing the plasma levels of IL-6, CRP, and fibrinogen in hypertensive patients with severe periodontitis.
Conclusion
The concept of focal infection, while shifting in and out of favour as a pathogenic mechanism, has always been recognized as being potentially causal in bacterial endocarditis. Most recently, intense attention has focused on ‘‘oral sepsis’’ and its relation to the causology of conditions such as osteopenia, diabetes, cardiovascular disease (CVD), and pre-term low-birth-weight infants. It is interesting to note that the classical risk factors of cardiovascular disease (hypertension, hypercholestrolaemia, cigarette smoking) can only account for one-half to one-thirds of the variation in the incidence of CVD cases. Thus it is likely that other factors may contribute to the pathogenesis of CVD. Several studies have pointed to a possible relationship between chronic oral infections and the pathogenesis of atherosclerosis and diseases associated with thromboembolic events such as myocardial infarction and stroke.
Bibliography
1. Beck JD, Garcia RG, Heiss G, Volconos P, Offenbacher S. Periodontal disease and cardiovascular disease. J Periodontol 1996; 67(suppl): 1123–1137.
2. Stein M, Kuch B, Conrads G, Fickl S, Chrobot J, Schulz S, Ocklenburg C, Smeets R. Clinical periodontal and microbiologic parameters in patients with acute myocardial infarction. J Periodontol 2009; 80(10): 1581-1589.
3. Ross R. Atherosclerosis- an inflammatory disease. N Engl J Med 1999; 340: 115-126.
4. Genco RJ, Offenbacher S, Beck J, Rees T. Cardiovascular diseases and oral infections. In: Rose LF, Genco RJ, Mealey BR, Cohen DW. Periodontal Medicine,1st Edition, 2000, B. C. Decker Inc.
5. Chun Y-HP, Chun K-RJ, Olguin D’A, Wang H-L. Biological foundation for periodontitis as a potential risk factor for atherosclerosis. J Periodont Res 2005; 40: 87–95.
6. Khlagatian M, Nassar H, Chou H-H, Gibson FC, Genco CA. Fimbriae- dependent activation of cell adhesion molecule expression in P.gingivalis-infected endothelial cells. Infect Immun 2002;70: 257-267.
7. Kuramitsu HK, Qi M, Kang I, Chen W. Role for periodontal bacteria in cardiovascular diseases. Ann Periodontol 2001; 6: 41-47.
8. Sharma A, Novak EK, Sojar HT, Swank RT, Kuramitsu HK, Genco RJ. Porphyromonas gingivalis platelet aggregation activity: outer membrane vesicles are potent activator of murine platelets. Oral Microbiol Immunol 2000: 15: 393–396.
9. Mattila KJ, Valle M, Nieminen MS,et al. Dental infections and coronary atherosclerosis. Atherosclerosis 1993;103: 205-211.
10. Renvert S, Lindahl C, Ann-Marie Roos-Jansaker,and Lessemi J. Short-Term Effects of an Anti- Inflammatory Treatment on Clinical Parameters and Serum Levels of C-Reactive Protein and Proinflammatory Cytokines in Subjects With Periodontitis. J Periodontol 2009;80:892-900.
11. Vidal F, Figueredo CMS, Cordovil I, Fischer RG. Periodontal therapy reduces plasma levels of Interleukin-6, C-reactive protein, and fibrinogen in patients with severe periodontitis and refractory arterial hypertension. J Periodontol 2009; 80(5): 786-791. |