Key Message:
Dentinogenesis imperfecta commonly known as opalescent dentin is one of the most common hereditary diseases of dentin affecting either or both dentitions. The aims of treatment are to restore function , aesthetics and protect teeth from wear.
Introduction:
Human dentition is subjected to considerable variations in size, form and number of teeth as well as in structure of dental tissues. Disorders of teeth may be hereditary. Dentinogenesis imperfect (DGI) is one of them.
Dentinogenesis imperfecta is a localized mesodermal dysplasia affecting both the primary and permanent dentitions.[1] It was first recognized by Barret in 1882.[2] The term was coined by Robert and Schour in 1939.[3] The first published report describing the disorder as an enamel defect was reported by Talbot as quoted by Witkop. The term ‘ hereditary opalescent dentin ‘ was first used by Skillen , Finn and Hodges to describe the brown translucent that have an opalescent sheen and are lacking in pulp chambers.[4].
Classification:
The most familiar classification system of this hereditary disorder is that formulated by Shields in 1973. This categorization discriminates three types of dentinogenesis imperfecta. Type I, Type II, Type III. [5]
The Shields' system is increasingly out of date as it does not account for the molecular aetiologies of the hereditary dentin defects elucidated so far. Hence the revised classification
Dentinogenesis Imperfecta I: dentinogenesis imperfecta without osteogenesis imperfecta. This corresponds to dentinogenesis imperfect type II of shield classification.
Dentinogenesis Imperfecta II: dentinogenesis imperfecta brandywine type.This corresponds to dentinogenesis imperfecta type III of shield classification.
There is no substitute in the present classisfication for the category designated as DGI type I of the previous Shields classification.
Genetic Variation
As DGI is inherited in an autosomal dominant fashion, there is a 50% chance that a child born to an affected parent will themselves be affected. This also indicates uniform gene expression[6] and complete penetrance for the gene in the family.[7].
Aetiology:
Mutations in the genes encoding the major protein constituents of dentine seem to underlie most hereditary dentine defects.
The only mutations causative of DGI, are found in the dentine sialophosphoprotein gene (DSPP), suggesting that these conditions are indeed allelic. DSPP is located within human chromosome 4q22.1 and consists of 5 exons spanning approximately 8343 bp.[8] Three distinct protein products are formed from the initially translated polypeptide: dentine sialoprotein (DSP), dentine glycoprotein (DGP) and dentine phosphoprotein (DPP).
As a consequence of the repetitive nature of that region of DSPP which encodes DPP (exon 5), all of the DGI- causing mutations that were initially detected were located in the DSP coding region and were composed of mis-sense, non-sense and splicing mutations. A comprehensive analyses of DSPP have demonstrated that DGI can result from mutations in that region of the gene which encodes DPP.[11] These mutations are exclusively deletions that lead to frame-shifts which change tandem hydrophilic serine-serine-aspartic amino acid repeats to long stretches of hydrophobic residues rich in valine, alanine and isoleucine .Moreover, a broad genotype- phenotype correlation has been reported for the DPP mutations with the most 5' mutations, which result in the longest sequences of hydrophobic amino acids, underlying and the more 3' mutations underlying DGI-II/ III. [9,10]
Clinical Features
Extraoral features
Short stature
Blue sclera.
Sensorineural hearing loss has also been reported as a rare feature of the DGI II.[11]
 | Fig 1
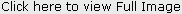 |
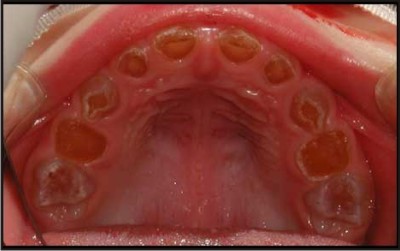
Intraoral features Fig 1
In both dentitions tooth colour - may vary from normal to amber, grey or purple to bluish translucent discolouration.
Excessive attrition and tooth wear is seen.
Abscess formation can be there
Tooth mobility is shown invariably
There is generally early loss of teeth.
The tooth enamel may have sheared off leaving dentine exposed; in such cases the exposed dentine often has a hard glassy appearance due to sclerosis. For this reason, patients rarely complain of sensitivity.
Histological Features:
DGI has less numerous crystallites, high Ca/P ratio and carbonated apatite [12,13]
 | Fig 2
|
Radiographic Features Fig 2
Radiographs should reveal normal enamel and dentine radiodensity, however, the enamel may already be lost with only dentine remaining.
Crowns may appear bulbous with marked cervical constriction.
Pulp chambers and canals may be normal, contain pulp stones or, more often, be partially or totally obliterated.
Sharp conical roots with apical constrictions or rootless teeth.
There may be numerous periapical radiolucencies in non-carious teeth
Pulpal remnants parallel to the cemento-enamel junction are seen in permanent teeth.
Diagnosis
Diagnosis is based on history, clinical examination and radiographic features. Clinical examination and radiographic follow up at various ages is recommended to determine diagnosis. [14]
Differential Diagnosis
Included in the differential diagnosis are conditions that have similar clinical or radiographic features to DGI.
a) Exposure of underlying dentine
Hypocalcified forms of amelogenesis imperfecta initially develop normal enamel thickness but the poorly calcified enamel is soft and friable and is rapidly lost by attrition leaving dentine cores. Unlike DGI the teeth are usually sensitive and on radiographs enamel is less radio-dense than dentine . [15] Pulp chamber and root canals are usually not sclerosed.
b) Intrinsic discolouration
Congenital erythropoietic porphyria is a condition resulting from an inborn error of porphyrin metabolism. This deficiency leads to haemolytic anaemia, photosensitivity, blistering of the skin, and deposition of red-brown pigments in the bones and teeth. [16]
Rhesus incompatibility- The discolouration which ranges from yellow through to green, brown and grey to black is usually found at the necks of teeth and the enamel hypoplasias are usually located in the coronal third of the teeth.[17]
Tetracyclines have the ability to chelate calcium ions and to be incorporated into developing teeth, cartilage and bone, resulting in discolouration of both the primary and permanent dentitions. This permanent discolouration varies from yellow or grey to brown depending on the dose or the type of the drug received in relation to body weight . [18]
c) Mobility leading to early tooth loss
Other causes of early loss of teeth as in DGI include:
hypophosphatasia, immunological deficiencies e.g. severe congenital neutropenia (Kostmann's disease), cyclic neutropenia, Chediak-Hegashi syndrome, neutropenias, histiocytosis X, Papillon-Lefevre syndrome and leucocyte adhesion deficiency syndrome . With the exception of hypophosphatasia, all of these conditions have an underlying immunological defect which makes those with these conditions susceptible to periodontal breakdown. Mobility of teeth in those with hypophosphatasia however is due to aplasia or marked hypoplasia of cementum.
d)Vitamin D-dependent rickets and vitamin D-resistant rickets
They have clinical and radiographic features of DGI. Vitamin D-dependent rickets is characterised by yellowish to brown enamel, chronic periodontal disease, large quadrangular pulp chambers and short roots. Features of vitamin D-resistant rickets include attrition and exposure of abnormally formed dentine of primary teeth and abscessed non-carious primary or permanent teeth.
Management
The aims of treatment are to remove sources of infection, restore function,aesthetics and protect posterior teeth from wear and maintain the occlusal vertical dimension. Treatment varies according to the age of the patient, severity of the problem and the presenting complaint.
Primary Dentition
Stainless steel crowns on the molars may be used to prevent tooth wear and maintain the occlusal vertical dimension.
The aesthetics may be improved using composite facings or composite strip crowns[19] .
If, however the child presents late, the teeth may have undergone attrition to the level of the gingivae and the only treatment option then is to provide over-dentures.[20] Children usually adapt well to over-dentures but they need to be reviewed regularly and dentures remade, as the child grows. If abscesses develop, pulp therapy is not successful and removal of the affected teeth is required. In younger children, where co-operation is limited, or the level of treatment required is extensive, a general anaesthetic may be required to facilitate treatment. In some cases, the parents or child may not be concerned with aesthetics and may request removal of sources of pain or infection only.
Fissure sealeants may be given to erupted molars to protect them from caries.[21]
Permanent Dentition
As the permanent dentition erupts, it should be closely monitored in relation to the rate of tooth wear with intervention only if necessary.
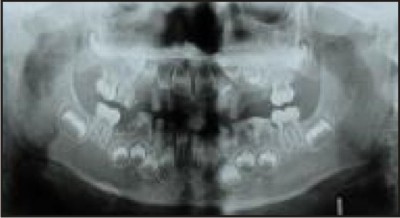
Cast occlusal onlays on the first permanent molars and eventually the premolars, help to minimise tooth wear and maintain the occlusal vertical dimension . Fig 3
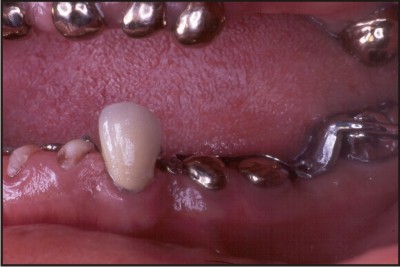 | Fig 3
|
The emphasis should be on minimal tooth preparation until the child reaches adulthood. At this point, if clinically indicated, a full mouth rehabilitation may be considered with full crowns and partial dentures. Teeth with short thin roots and marked cervical constrictions however are often unfavourable for crowns . Obliteration of the pulp chambers and root canals in teeth that develop abscesses makes endodontic therapy difficult if not impossible.
Successful conventional endodontic therapy can be done. If conventional therapy is not an option, periapical curettage and retrograde root filling is another possible alternative, but is not recommended for teeth with short roots.
Some patients present with severe tooth wear in the permanent dentition. As in the primary dentition, one of the options is over-dentures. As the denture bear area is less so there are chances of midline fracture , due to flexural fatigue and impact force.To avoid this, denture base can be reinforced with silanized glass fibres. [22]
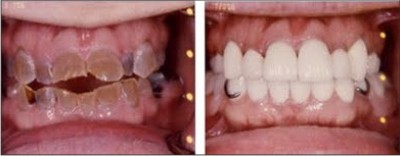 | Fig 4
|
Veneers [FIG4] can be considered as a possible treatment option in case of toothwear and to improve aesthetics in anterior teeth. direct and indirect composite veneers are useful as an interim measure until the child is older in order to manage anterior aesthetics. porcelain veneers can be replaced on the teeth in permanent dentition
Those with have mobile teeth due to very short roots and as a result tend to lose teeth early in the primary and permanent dentition. Until growth is complete, the treatment of choice for thr replacement of missing teeth is dentures.
Dental implants may be considered when growth is complete at about 18 years of age. A canine protected occlusion may be considered to decrease the lateral forces on posterior dentition. [23]
Maxillo-mandibular atrophy is a consequence of no or rudimentary root development and early tooth loss. Ridge augmentation prior to implants is often required .
Exposed dentine is more susceptible to tooth decay than enamel. For all patients, regular dental checkups and prevention of tooth decay in the form of oral hygiene instruction, dietary advice and appropriate use of fluoride is essential. Early diagnosis and regular dental care can prevent premature tooth-loss due to short or absent roots and spontaneous abscess formation , so a good aesthetics and function can be obtained.
References:
1. Witkop CJ, RaoS. Inherited defects in tooth structure . Baltimore, Williams and Wilkins. 1971; 7:153-84.
2. Bhandari S, Pannu K. Dentinogenesis imperfecta: a review and case report of a family over four generations. Indian J Dent Res. 2008;19:357-61.
3. Kamboj M, Chandra A. Dentinogenesis imperfecta type II: an affected family saga. J Oral Sci. 2007 ;49:241-4.
4. Subramaniam P, Mathew S, Sugnani SN. Dentinogenesis imperfecta: a case report. J Indian Soc Pedod Prev Dent. 2008 ;26:85-7.
5. Barron MJ, McDonnell ST, Mackie I, Dixon MJ. Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia. Orphanet J Rare Dis. 2008 ;3:31.
6. Bixler D, Conneally PM, Christen AG. Dentinogenesis imperfecta: genetic variations in a six-generation family. J Dent Res. 1969;48:1196-9.
7. Crosby AH, Scherpbier-Heddema T, Wijmenga C, Altherr MR, Murray JC, Buetow KH, Dixon MJ. Genetic mapping of the dentinogenesis imperfecta type II locus. Am J Hum Genet. 1995 ;57:832-9.
8. Bai H, Agula H, Wu Q, et al .A novel DSPP mutation causes dentinogenesis imperfecta type II in a large Mongolian family. BMC Med Genet. 2010 ;11:23
9. McKnight DA, Simmer JP, Hart PS, Et Al. Overlapping DSPP Mutations Cause Dentin Dysplasia And Dentinogenesis Imperfecta. J Dent Res. 2008 ;87:1108-11.
10. Zhang X, Chen L, Liu J, Zhao Z.Et Al. A Novel DSPP Mutation Is Associated With Type II Dentinogenesis Imperfecta In A Chinese Family. BMC Med Genet. 2007 ;8:52
11. ShaferWG, HineMK, LevyBM. Disturbances Of Development And Growth. In: Rajendran R, Sivapathasundram B,editors. Shafer’s Textbook Of Oral Pathology. Elsevier: A Division Of Reed Elsevier India Private Limited ;2006.
12. Nanci A.Dentin-Pulp Complex. In: Nanci A. editors. Ten Cate's Oral Histology: Development,Structure and Function. 7th ed. St. Louis, Missouri, USA: Mosby Elsevier; 2008.
13. Kerebel B, Daculsi G, Menanteau J, Kerebel LM. The inorganic phase in dentinogenesis imperfect. J Dent Res. 1981;60:1655-60.
14. Barron MJ, McDonnell ST, Mackie I, Dixon MJ. Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia. Orphanet J Rare Dis. 2008 ;3:31.
15. Witkop CJ Jr: Amelogenesis imperfecta, dentinogenesis imperfecta and dentin dysplasia revisited: problems in classification. J Oral Pathol 1988 , 17:547-53.
16. Fayle SA, Pollard MA: Congenital erythropoietic porphyria – oral manifestations and dental treatment in childhood: a case report. Quintessence Int 1994 , 25:551-4.
17. Pindborg JJ: Aetiology of developmental enamel defects not related to fluorosis. Int Dent J 1982 , 32:123-34.
18. Sanchez AR, Rogers RS 3rd, Sheridan PJ: Tetracycline and other tetracycline-derivative staining of the teeth and oral cavity. Int J Dermatol 2004 , 43:709-15.
19. Tahmassebi JF, Day PF, Toumba KJ, Andreadis GA: Paediatric dentistry in the new millennium: 6. Dental anomalies in children. Dent Update 2003 , 30:534-40.
20. Dhaliwal H, Mckaig S. Dentinogenesis Imperfecta – Clinical Presentation And Management. Dent update 2010;37:364-71.
21. Delgado Ac, Alarcon Ja. Dentinogenesis Imperfecta: The Importance Of Early Treatmnet. Quint Int 2008,39:257-63.
22. Henke DA, Fridrich TA. Occlusal Rehabilitation Of The Patient With Dentinogenesis Imperfecta: A Clinical Report. J Prosthet Dent 1999;81:503-6.
23. Goud A. Full Mouth Rehabilitation Of A Patient Suffering From Dentinogenesis Imperfecta. Dent Practice 2010;9:34-6. |