|
|
|
| Laugier-hunziker Syndrome: A Review |
Ankur Bhargava 1 , Sonal Saigal 2
1 Senior lecturer, Department Of Oral Pathology - Government Dental College, Raipur (Chhattisgarh), India
2 Senior lecturer, Department Of Oral Pathology - Government Dental College, Raipur (Chhattisgarh), India
|
| Address For Correspondence |
Dr. Sonal Saigal
291- A Block,
Chitrakut Nagar,
Bhuwana Extension,
Udaipur (Rajasthan).
E-mail: sonal_ankur@rediffmail.com |
| Abstract |
| Pigmentation is frequently encountered in the oral mucosa. Focal lesions usually need an in-depth examination to exclude a melanoma, while diffuse lesions often have no specific histological features and do not generate prognostic perplexity. However, diagnosis of these lesions is important because they could be a sign of diseases with systemic implications such as Peutz-Jeghers syndrome or adrenal insufficiency. Laugier-Hunziker syndrome (LHS) is a rare acquired macular hyperpigmentation of oral mucosa and lips frequently associated with longitudinal pigmentation of the nails. The pathogenesis is unknown, but no systemic involvement or malignant predisposition has been described, so the correct clinical identification avoids the need for detailed and potentially hazardous investigations and treatment. |
|
| Keywords |
| Pigmentation, Laugier-Hunziker syndrome, Peutz-Jeghers syndrome. |
|
| Full Text |
INTRODUCTION
Pigmentation is both the normal and abnormal discoloration of oral mucous membrane. It has multifactorial etiology. Most of the pigmentation is physiologic but sometimes it can be a precursor of severe diseases. A practical approach in a clinical situation is to examine whether the pigmentation presents as focal or as diffuse lesions (Table)1.
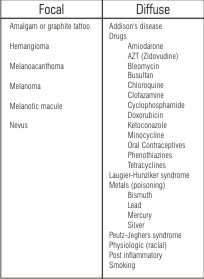 | Table. Causes of focal and diffuse oral pigmentation
 |
Sometimes the clinical presentation of pigmentation together with a thorough medical and family history, as well as history of onset, duration, and progression of the pigmentation, guides us to make a suggestive diagnosis. The clinical behavior of focal oral pigmented lesions ranges from benign, requiring no treatment, to highly malignant. Therefore, a biopsy is usually required for accurate diagnosis of a focal pigmented lesion2.
Oral pigmentation caused by systemic diseases is usually diffuse and multifocal and has no specific histologic features. Although a solitary pigmented lesion may cause more suspicion, it should be kept in mind that diffuse oral pigmentation may be the first manifestation of an underlying systemic disease1. It has been associated with a variety of endogenous and exogenous etiologic factors. Most pigmentation is caused by five primary pigments. These include: melanin, melanoid, oxyhemoglobin, reduced haemoglobin, and carotene. Others are caused by bilirubin and iron. Melanin pigment irregularities and color changes of the oral tissues could provide significant diagnostic evidence of both local and systemic disease3.
Laugier-Hunziker pigmentation (LHP) is an acquired disorder of hypermelanosis and involves the lips, oral mucosa, nails and acral areas in varying combinations that was first described by Laugier and Hunziker in 19704. LHP may resemble various disorders characterized by mucocutaneous pigmentation. Differentiation from these disorders is essential as a few of them imply associated systemic disorders. LHP was initially thought to be restricted to European countries, now it has been reported from many parts of world, including India5,6,7.
PATHOGENESIS
The cause of this rare disease is yet unknown. Ultrastructural studies reveal an increase in the number and size of mature melanosomes located in the cytoplasm of basal keratinocytes and dermal melanophages8. Some authors suggest that functional alteration of the melanocytes in the form of an increase in the synthesis of melanosomes and their subsequent transport to the basal layer cells may give rise to hyperpigmented lesions1,9.
CLINICAL FEATURES
The onset of the condition is usually in early or mid adult life; the mean age of patients is 52.3 Women are affected more often than men, and most reported cases have been in whites. Neither familial association nor any associated systemic diseases have been reported, to date. LHS is mainly a disease of Caucasians and most of the reported cases have been from the European countries8.
The pigmentation consists of slate to dark brown lenticular (lens-shaped) or linear macules, solitary or confluent, with well-defined or indistinct margins. The lesions are located most often on the buccal mucosa and on the lips1. Other locations include the hard and soft palate, the tongue, and, more rarely, the gingiva or floor of the mouth. Pigmented macules may also occur on the neck, thorax, abdomen, perineal and perianal region, sclera and esophagus10,11.
Nail involvement is seen in about 60% of the patients; it consists of pigmented longitudinal bands of varying width and intensity in 1 or more of the fingernails and/or toenails without nail dystrophy. Baran et al. proposed three types of nail pigmentation in LHS: (i) isolated longitudinal streaks of varying degrees of pigmentation1 to 2 mm in width, (ii) 2 to 3 mm double longitudinal streaks, and (iii) homogenous pigmentation of the radial or ulnar half of the nail12. Rarely, the pigmentation may spread from the proximal nail fold into the surrounding skin, a phenomenon known as Hutchinson's sign, which is associated with malignancy but also with benign lesions such as recurring junctional nevus. The hyperpigmentation associated with LHS occurs spontaneously and may progress slowly or remain stable. There are neither systemic findings nor genetic factors associated with the syndrome1.
HISTOPATHOLOGY
Histologically, the pigmented lesions in LHS show epithelial acanthosis and increased melanin pigmentation in the basal layer of the epithelium. Melanophages are seen in the upper lamina propria and dermis. Melanocytes are normal in number, morphology, and distribution1.
Epidermal and epithelial basal layer pigmentation is common in skin and mucosal lesions, respectively, with pigment- laiden melanophages evident in the papillary dermis. One report describes abundant melanin “free” in the papillary dermis13.
Acanthosis of the epidermis is emphasized in several cases; saw tooth or elongation of rete ridges is noted in 2 cases14,15, whereas no elongation of epidermal rete ridges is described in other cases16,17.
Two recent reports demonstrated increased intraepidermal melanocytosis in the lesions of LHP, with one report also describing significant cellular atypia of intraepithelial melanocytes from a macular lesion on a sun-exposed area18,19.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of LHP includes Peutz- Jeghers syndrome (PJS), LAMB syndrome and LEOPARD syndrome. Intraepidermal melanocytic hyperplasia has been identified in the pigmented macules of all these syndromes.
LAMB syndrome is characterized by lentigines of the skin and mucosa, atrial and mucocutaneous myxomas, and multiple blue nevi, while LEOPARD syndrome is characterized by lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and deafness4.
The Peutz-Jeghers syndrome (PJS) shares most clinical features with LHS and must be ruled out in case of diffused oral pigmentation because it may be associated with an increased incidence of gastrointestinal as well as genital and mammary tumors. PJS is an autosomal dominant inherited disease with a high degree of penetrance characterized by intestinal polyposis and melanotic macules, particularly of the face and mouth20.
The differential diagnosis between PJS and LHS may be hampered by overlapping clinical and histological features. However, some characteristics may help to differentiate the two syndromes: the appearance of the lesions in infancy or early childhood and the presence of family hyperpigmentation or intestinal polyposis, or pigmentation also on the face, hands, and feet, suggest PJS, while LHS can be assumed when both oral and nail pigmentations are present21.
Other differential diagnosis include: Addison's disease is an endocrine disorder due to an insufficient production of cortisol and aldosterone that can present with diffuse hyperpigmentation of the skin and mucous membranes. The oral manifestations are primarily due to an increased level of circulating adrenocorticotropic hormone (ACTH) and may be the first sign of the disease, so the exact interpretation of the lesions is mandatory for prompt diagnosis and to institute appropriate therapeutic strategies. No clinical signs of systemic symptoms such as fatigue, weight loss, and cardiovascular or gastrointestinal disorders were found in our patient, and her plasma levels of cortisol and ACTH were normal22.
Albright’s syndrome is genetic disorder of bones, skin pigmentation, and hormonal problems with premature sexual development. Pigmentary changes are not pathognomonic but may include irregular often unilateral truncal pigmentation (caf´e-au-lait macules),macular lip, and genital pigmentation. No nail pigmentation has been reported. The syndrome manifests in childhood and this excludes the possibility of such pathology in our patient22.
Diffuse oral pigmentation may also be associated with systemic intake of drugs such as tetracyclines, antimalarials, amiodarone, chemotherapeutic agents, oral contraceptives, phenothiazines, azidothymidine, and ketoconazole. A correct diagnosis will resolve the drug-induced oral mucosal pigmentation following the suspension of the causative drug23.
Smoking may produce oral pigmentation called smoker's melanosis, is usually confined to the anterior attached gingival and not associated with pigmentation in other parts of the body24.
TREATMENT AND PROGNOSIS:
No literature reports have described a progression of LHS to oral cancer, and therefore all cases must be simply followed up without any specific treatment25. The reported treatment options include Q-switched Alexandrite laser, Q-switched Nd-Yag laser, and cryosurgery26,27 .
In conclusion, LHS rare syndrome probably not well known among general dentists. Dentists should therefore be familiar with the Laugier-Hunziker syndrome as a benign condition not requiring treatment. When a patient presents with diffuse oral pigmentation, detailed history taking and thorough clinical examination including fingernails will establish the diagnosis and exclude local or systemic diseases requiring medical management.
REFERENCES
1. Siponen M, Tuula S, Oulu F. Idiopathic lenticular mucocutaneous pigmentation (Laugier-Hunziker syndrome): A report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;96:288-92.
2. Eisen D. Disorders of pigmentation in the oral cavity. Clin Dermatol 2000;18:579-87.
3. Çiçek Y. The Normal and Pathological Pigmentation of Oral Mucous Membrane: A Review. J Contemp Dent Pract 2003;3:76-86.
4. Ajith C, Handa S. Laugier-Hunziker pigmentation. Indian J Dermatol Venereol Leprol 2005;71:354-6.
5. Lenane P, Sullivan DO, Keane CO, Loughlint SO. The Laugier- Hunziker syndrome. J Eur Acad Dermatol Venereol 2001;15:574-7.
6. Kanwar AJ, Kaur S, Kaur C, Thami GP. Laugier-Hunziker syndrome. J Dermatol 2001;28:54-7.
7. Ayoub N, Barete S, Bouaziz JD, Le Pelletier F, Frances C. Additional conjunctival and penile pigmentation in Laugier- Hunziker syndrome: a report of two cases. Int J Dermatol 2004;43:571-4.
8. Engin S, Hakan E, Doðan K, Nurper F, Zafer K, Gülhane T. Laugier-Hunziker sendromu: bir olgu sunumu. Gulhane Tip Dergisi 2006;48:104-6.
9. Veraldi S, Cavicchini S, Benelli C, Gasparini G. Laugier-Hunziker syndrome: a clinical, histopathologic, and ultrastructural study of four cases and review of the literature. J Am Acad Dermatol 1991;25:632-6.
10. Yamamato O, Yoshinaga K, Asahi M, Murata I. A Laugier-Hunziker syndrome associated with esophageal melanocytosis. Dermatol 1999;199:162-4.
11. Mowad CM, Shrager J, Elenitsas R. Oral pigmentation representing Laugier-Hunziker syndrome. Cutis 1997;60:37-9.
12. Baran R, Barriere H. Longitudinal melanonychia with spreading pigmentation in Laugier-Hunziker syndrome: a report of two cases. Br J Dermatol 1986;115:707-10.
13. Harris A, Mace MC, Burge SM. An unusual case of the Laugier-Hunziker syndrome. Br J Dermatol 1994;131:84-9.
14. Koch SE, LeBoit PE, Odom RB. Laugier-Hunziker syndrome. J Am Acad Dermatol 1987;16:431-4.
15. Seoane Leston JM, Vasquez Garcia J, Cazenave Jimenez AM, de la Cruz Mera A, Aguado Santos A. Syndrome de Laugier-Hunziker: etude Clinique et anatomopathologique. Presentation de treize cas. Rev Stomatol Chir Maxillofac 1998;98:44-8.
16. Revuz J, Clerici T. Penile melanosis. J Am Acad Dermatol 1989;20:567- 70.
17. Kemmet D, Ellis J, Spencer MJ, Hunter JA. The Laugier-Hunziker syndrome- a clinical review of six cases. Clin Exp Dermatol 1990;15:111-4.
18. Moore RT, Chae KA, Rhodes AR. Laugier and Hunziker pigmentation: A lentiginous proliferation of melanocytes. J Am Acad Dermatol 2004;50:70-4.
19. Koch SE, Le Boit PE, Odom RB. Laugier-Hunziker syndrome. J Am Acad Dermatol 1987;16:431-4.
20. Rubio A, Ram´?rez A, Angeles A, Uscanga L. Peutz-Jeghers syndrome. Revista de Gastroenterolog ´?a de M´exico 2005;70:291-5.
21. . Sardana K, Mishra D, Garg V. Laugier-Hunziker syndrome. Indian Pediatrics 2006;43:998-1000.
22. Nieman LK, Chanco M L. "Addison's disease". Clinics Dermatol 2006;24:276-80.
23. Dereure O. Drug-induced skin pigmentation epidemiology, diagnosis and treatment. American Journal of Clinical Dermatology 2001;2:253-62.
24. Mirbod SM, Ahing S. Tobacco-associated lesions of the oral cavity: part I. Nonmalignant lesions. J Canadian Dental Asso 2000;66:252-6.
25. Lucio M, IvanaG, Fabio C, Cosimo M, BeatriceR. Laugier-Hunziker Syndrome: An uncommon cause of oral pigmentation and a review of the literature. Internation J Dentistry 2010;1:1-4.
26. Papadavid E, Walker NP. Q-switched Alexandrite laser in the treatment of pigmented macules in Laugier- Hunziker syndrome. J Eur Acad Dermatol Venereol 2001;15:468- 9.
27. Sheridan AT, Dawber RP. Laugier- Hunziker syndrome: treatment with cryosurgery. J Eur Acad Dermatol Venereol 1999;13:146-8. |
|
|
|
|
|
|