INTRODUCTION
Ectodermal dysplasia (ED) is a hereditary disorder that can affect several ectodermal structures. These structures may include: skin, hair, nails, teeth, nerve cells, sweat glands, parts of the eye and ear, and parts of other organs. Thurman published the first report of a patient with ED in 1848, but the term was not coined until1929 by Weech. 1,2,3
ED syndromes have been described as a group of disorders of morphogenesis displaying two or more of the following signs and symptoms.1,2,3
1. Trichondysplasia (abnormal hair)
2. Abnormal dentition
3. Onchondysplasia (abnormal nails)
4. Dyshidrosis (abnormal or missing
sweat glands)
Lamartine in 2003 has described various well defined ectodermal dysplasia as Hypohidrotic (anhidrotic),Hidrotic (Clouston’syndrome), Ectrodactyly-ectodermal dysplasia-cleft syndrome (EEC),Rapp- Hodgkin Syndrome,Hay-Wells syndrome or an kyloblepharonectodermal dysplasia.Usually the ectodermal dysplasia is divided into two types based on the number and function of sweat glands as mentioned below4:
1. Hypohidrotic (anhidrotic)
Ectodermal Dysplasia
(Christ-Siemens- TourineSyndrome)
2. Hidrotic Ectodermal Dysplasia
(CloustonSyndrome)
Hypohidrotic (anhidrotic) Ectodermal Dysplasia (Christ-Siemens- Tourine Syndrome)
Hypohidrotic (anhidrotic) Ectodermal Dysplasia (Christ-Siemens- Tourine Syndrome)
The most reported ED syndrome is x-linked hypohidrotic (anhidrotic) ED (Christ-Siemens-Touraine syndrome) which affects one to seven Individuals per 10,000 with males afflicted more frequently than, females1,2,5,6-9.
Patients generally have prominent supraorbital ridges, frontal bossing, and a saddle nose. The maxilla may be under developed and the lips are thick and prominent. The nose may appear pinched and the aleque nasi appears hypoplastic. The patient may resemble like an old edentulous person 8,9.
The skin is usually dry, scaly, and easily irritated as a result of poorly developed or absent sebaceous glands. Sweat glands may also be absent or few in number or non-functioningwhich may result in increased body temperature. Scalp hair may be absent, sparse, very fine pigmented, or abnormal in texture. Eyebrows, eye lashes, and other body hair may also be sparse or absent. When hairs are present, they may be fragile, dry, and generally disorderly because of the lack of sebaceous glands. Finger and toe nails are usually normal 9.
Orofacial characteristics of this syndrome include anodontia or hypodontia, hypoplastic conical teeth, underdevelopment of the alveolar ridges, frontal bossing, a depressed nasal bridge, protuberant lips, and hypotrichosis 10,11. Teeth in the permanent dentition are frequently small, conical, tapered (peg shaped), and widely spaced. Lack of alveolar growth may be associated with this condition and frequently results in increased inter-occlusal distance which allows optimum artificial tooth placement 12. Patients may present with a marked mandibular protrusion. Depending on the severity of the condition, various prosthodontic treatments are available to improve appearance, mastication, and speech 13.
Children usually have a normal mentality and life expectancy, and their facial appearance warrants professional concern for their emotional well being and social progress8,14. Tanner 15 states ectodermal dysplasia with an abnormal appearance may affect normal social and psychological development in young patients. Functional needs also must be considered since the difficulty these children experience in masticating may cause nutritional problems 8, 14. Therefore, dental care for such patient is important.
Hidrotic Ectodermal Dysplasia (Clouston Syndrome)
Here the clinical features include nail dystrophy, hair defects and palmo-plantar dyskeratoris. The patients have normal facies, normal sweating and no specific defect is seen.
Anhidrotic ectodermal dysplasia (EDA) is a rare disorder commonly transmitted as an X-linked reccssive disorder. However, rarely autosomal recessive and autosomal dominant inheritance have also been seen. This syndrome manifests as a triad of defects partial or complete absence of sweat glands anomalous dentition and hypotrichosis.16In view of the rarity of this entity, we report two cases of anhidrotic ectodermal dysplasia.
Case reports:
Case 1:
A 12-year old male child reported to our hospital with complaint of lack of esthetics and difficulty in mastication. Patient also complained of fever on and off. Fever used to be high grade and more so during summer and would subside with medication.The patient had history ofparchment like skin sheet at birth
Clinicalexamination of the patient revealed a fine, sparse scalp hair on scalp while hair were absent on eyebrows, and eyelashes. There was frontalbossing, depressed nasal bridge and malar hypoplasia. Wrinkledhypo-pigmented skin around periorbital region was noted. Skin was rough and dry with no sweat on it (Figure 1).
 | Fig 1
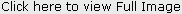 |
The teeth were reduced in number and conicalin shape in upper jaw and anodontia in lower jaw (Figure 2), the alveolar ridges were underdeveloped, and the lips were prominent.
 | Fig 2: Oral examinationof Case No. 1, showing teeth were reduced in number and conical in shape in upper jaw and anodontia in lower jaw.
|
The treatment option preferred was of a removable partial denture for the missing maxillary teeth and complete denture for edentulous mandibular arch. The peg shaped teeth were modified with composite resin which helped in enhanced retention ofupper denture. The dentures were delivered and the patient was instructed on the maintenance of oral hygiene and dentures. Recall appointments were done for adjustments.(Figure 3)
Case 2:
An 11 year old young female, presented to our hospital for evaluation of teeth abnormalities. She had a history of recurrent high fevers duringinfancy, and the patient's mother reported that herdaughter is not able to sweat she had to apply someprecautions to protect her from overheating duringwarm weather and physical exertion.
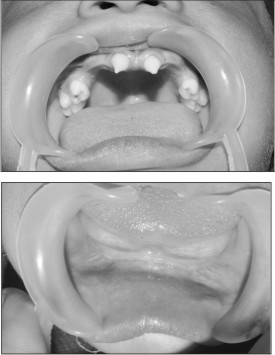
Other family members (including first degree cousins) wereaffected by hypohidrosis and dental abnormalities. Her mother’s medical history included four death births. Her family pedigree is represented at Figure 4.
 | Fig 4: Pedigree of Case No.2
|
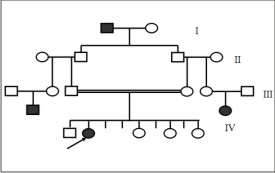
Clinical examination of the patient revealed a fine, sparsescalp hair, eyebrows, and eyelashes. The patient’sskin was smooth and dry. Mild periorbital andperioral hyperpigmentation were evident. Additional facialfeatures included frontal bossing, prominentsupraorbital ridges, and midfacial hypoplasia with adepressed nasal root and bridge. (Figure 5)
 | Fig 5: Clinical picture of Case No. 2, revealing fine, sparse scalp hair, eyebrows, and eyelashes.
|
The patient hadoligodontia. The teeth were reduced in number andconical in shape.(Figure 6)
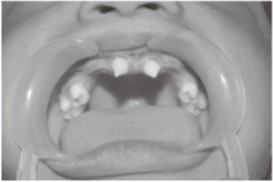 | Fig 6: Oral examinationof Case No. 2, showing oligodontia. The teeth were reduced in number andconical in shape.
|
OPG revealed complete absence of some permanent tooth germs andunderdeveloped maxillary and mandibular alveolarridges. (Figure 7)
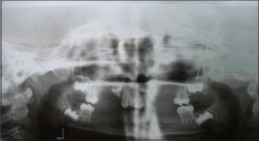 | Fig 7: OPG of Case No. 2, showing complete absence of some permanent tooth germs andunderdeveloped maxillary and mandibular alveolarridges.
|
The treatment option preferred was of a removable partial denture for the missing teeth.The peg shaped teeth were modified with composite resin which helped in enhanced retention of upper denture.
Discussion and review of literature:
Disease name and synonyms
Ectodermal dysplasia anhidrotic (EDA)
Anhidrotic ectodermal dysplasia
Hypohidrotic ectodermal dysplasia (HED)
Christ-Siemens-Touraine syndrome
Definition
Ectodermal dysplasias (EDs) are a heterogeneous group of disorders characterized by developmental dystrophies of ectodermal structures, such ashypodontia or anodontia.1,2,3
More than 190 clinically and genetically distinct hereditaryectodermal dysplasias have been catalogued 17.
Anhidrotic (hypohidrotic) ectodermal dysplasia (EDA) is the most common ED (80%); it is characterized by hypoplasia of hair, teeth and sweat glands. Since there is not a complete lack of sweat glands the term hypohidrotic is more adequate than anhidrotic. The anhidrotic (hypohidrotic) ectodermal dysplasia is ofteninherited as an X-linked disorder (XLEDA /Christ-Siemens- Touraine syndrome). 18X-linked anhidrotic ED was first described in 1848 by Thurnam and later in the 19th century by Darwin. Both autosomal dominant and autosomal recessive inheritance of clinically similar conditions have now been demonstrated and the molecular defects defined19.
Genetics of EDA
All the ectodermal dysplasias appear to be genetic in etiology.Of more than 190 EDs described, the molecular basis has beenelucidated for more than 30 of them20. A little less than a hundred years after Darw in’s discussion, the gene for EDA was the first Xchromosomal gene whose map position was suggested, based on the occurrence of an X; auto some translocation in a female patient (“Anly”) with the disease phenotype. Later linkage studies confirmed the suggested position of the gene at Xq12-q13.1 (XLHED-gene) 21. The localisation of the gene has permitted more accurate diagnostics of the disorder, both in carrier females and in prenatal screening. The discoveries of disease genes and the identification of mutations in patients represent great progress in biomedicalscience. In order to help patients suffering from the diseases it is obviously not enough that the gene defects are known. It is necessary to understandthe context in which genes function and the details of the biological processes where they are involved.
The recent cloning of the gene has led to theidentification of a novel transmembrane protein”ectodysplasin” (TNF family ligand) and receptor “edar” (TNF receptor). This TNF ligand and receptorhave a developmental regulatory role and are tightly associated with epithelial-mesenchymal interactionsand signaling pathways that regulate ectodermal appendage formation and organogenesis during the initiation of development 22.
The next step in research, the analysis of thefunction of the genes and the effects of genemutations will be a challenge for researchers for many decades to come, and this can be expectedto result in the development of new therapeutic methods. It is quite possible that these kinds of observations of gene function and interaction may form the basis of new therapeutic methods in the future.
Epidemiology
The prevalence of EDA is unknown; however, the incidence in male is estimated at 1 in 100,000 birthsalthough the condition is usually overlooked in in fants23. This X-linkedrecessive disorder affects males and is inherited through female carriers. This carriers-incidence isprobably 17.3 in 100,000 women. The autosomal dominant and autosomal recessiveinheritance EDA is an extremely rare condition.24
Clinical Features
EDA is characterized by the triad of signs comprising sparse hair (atrichosis or hypotrichosis),abnormal or missing teeth (anodontia or hypodontia) and inability to sweat due to lack ofsweat glands (anhidrosis or hypohidrosis). The lackof teeth and the special appearance were reportedto be major concerns.1,2,3
Most patients with EDA have a normal life expectancy and normal intelligence. However, thelack of sweat glands may lead to hyperthermia,followed by brain damage or death in early infancy, if unrecognized. Thus an early diagnosis is important. Families with EDA should therefore beoffered genetic counselling. 1,2,3
Craniofacial structures
Considering how much osseous and dental tissue is missing, these patients have surprisingly normal facial structures. Already in 1936, Tannhauser stated that the characteristic deformity of the cranial bones of all affected patients is such that the resemblance among the patients is bigger than when compared with their own unaffected sib 23.
Clinically, the forehead appears square, with frontal bossing, and there is a prominent supra-orbitalridge. The nose has a depressed nasal bridge and is called a saddle nose. The midface is depressedand hypoplastic, giving it a “dished-in” appearance. The cheekbones are high and broad, although theyappear flat and depressed as well. The chin may be pointed and the lips everted and protuberant 25.
In non-treated patients with EDA, craniofacial deviations from the norm increased with advancing age 26 with a tendency toward a Class III pattern with anterior growth rotation 27. Cephalometric analysis and anthropometry studies have been performed. The quantitative findings show reduced facial dimensions, decreased lower facial height, variablepattern in facial widths, the maxilla has been relatively more retruded than the mandible, thenasal alar width and mouth width were significantly smaller28.
This remarkable variability in facial dimensions and harmony found in patients with ED probablycorresponds to the different kinds of dysplasia, with different expression of the interested genes 29.
Oral structures
Dental defects represent a core clinical feature of many EDs: anodontia, polydontia, dysplastic teeth, retained primary teeth, deficient enamel development (amelogenesis imperfecta), dentine deficiency (dentinogenesis imperfecta), and underdevelopment of the alveolar ridge. In some EDs, the number of erupted teeth is reduced, the spacing of the teeth disrupted, and the periodontium affected. Disturbance of the enamel matrix may occur, making the teeth more susceptible to caries and altering the shape of the teeth, leading to a pegged appearance and additional accessory cusps 30, 31.
Missing teeth or the delay in teething often starts to worry the parents and leads to the diagnosis of EDA in the second year of life. A dentist should not hesitate to radiographicallyexamine a patient whose teeth have not erupted bythe appropriate age in order to exclude EDA. Thescreening limit for the first tooth to erupt is 15months 32.
Besides the delay in teething, the teeth appear radiographically abnormal in shape and structure. The enamel layer is thin and the cervical area of the tooth is constricted. Enamel is rarely hypoplastic 23. If at that stage aplasia of several teeth is seen, the patient should be referredto a geneticist in a paediatric unit with a suspicionof EDA diagnosis. Tooth crowns are small and abnormal in shape. Upper incisors and cuspids are always conical or pointed. Taurodontism, frequently on the second deciduous molars, is a common feature .Not only the shape is abnormal, but also the number. A severe hypodontia is a universal feature amongst affectedindividuals. All lacked some deciduous teeth andpermanent teeth. The number varies from four to twenty. A few patients have congenital anodontia. There are generally more teeth in the maxilla than inthe mandible, although both jaws can be toothless. Most often the lower incisors and premolars are missing, followed by the upperpremolars and incisors. The upper cuspids and first upper and lower molars are formed.33
The edentulous EDA patients do not have anyalveolar processes either. In those patients with some natural teeth, there is a striking difference in the intra-oral heightand breadth of the bone. In areas where no teeth have developed, the alveolar bone is missing and the bone ridge is verythin in contrast to the normal alveolus surroundingan occasional tooth.34
Many patients complain of dry mucous membranes in mouth and nose. Reduced salivary secretion hasbeen spotted in some EDA patients 34. The oral mucous glands should be missing in the lips. Autopsy has also shownabsence of mucous glands in the pharynx, larynx, trachea and bronchi. This is in agreement with the susceptibility to respiratory infections. The other salivary glands are not described in literature.Analysis of the saliva has revealed a reduced buffer capacity and an increased number of bacterialcultures. Most affected individuals were susceptible to dental caries 33.
Hair, nails, skin and skintags
Abnormalities of hair are present in all affected individuals. Most individuals have sparse, fine,slowly growing scalp hair. Some individuals are completely bald by their middle teens, whereasother individuals have normal amounts of scalp hair, though it may exhibit an abnormal texture. Sparseeyebrows and eyelashes were always found. Most individuals show decreased body hair, pubic hair,and/or axillary hair, but these features are more variable. However, beard and moustache hair are normal. Electron microscopy of hairs from affected and unaffected individuals showed noabnormalities.
About half of the affected individuals exhibit mild fingernail abnormalities and nail dystrophy. Slownail growth and split nails are most often reported. A few individuals had a longitudinal ridging, thinningand superficial peeling. Nail problems occur more frequently in older individuals. This suggests thatthe nail beds are more susceptible to progressive injury with age. Toenails were generally normal.
Most individuals report dry skin. Affected individuals have a smooth, almost velvety skin texture. Theskin of patients also seems to be “thinner” than expected for age. Some infants may have apremature look because of the thin skin. Scaling in the neonate may form a clue to diagnosis.
Almost all affected relatives have decreased sweating, and many show heat intolerance. Some individuals only sweat in certain areas on theirbody. Common sites of sweating include palms,soles and axillae. Because ofthe reduced number of sweat glands, there is adanger of hyperthermia. In this way EDA has beenassociated with sudden infant death. Thehyperthermia may also lead to brain damage, and is probably the cause of the rare cases of EDAreported with mental retardation. Subcutaneous fat is often diminished and over one third of the boyshave abnormalities of the breast, including absent or accessory nipples.Episodes of hyperpyrexia and severe respiratory infections are life-threatening components in EDA.The delay in teething often leads to the diagnosis in childhood. After the first critical years of life the patients experience a general improvement inhealth. The lack of teeth is the most important factor in determining the quality of life in these patients,particularly in later life. They all suffer greatly from their abnormal facial and dental appearance.33
Differential Diagnosis
The differential diagnostic problem is the distinction of the autosomal recessive form of HED (AR-HED) from X-linked HED. AR-HED is considerably less common than XLHED. The clinical features arequite similar in both conditions but due to the different mode of inheritance AR-HED affects bothmales and females and the heterozygotes have no signs at all. For adequate genetic counselling it isthus important to recognize XLHED heterozygotes by dental examination and sweat tests. To distinguish between AR- and XL-forms, the HED diagnosis should be followed by careful family history for ectodermal manifestations both in maleand female and by tests for heterozygote identification. The findings of equally affected malesand females, as well as the presence of consanguinity, support an autosomalrecessive mode of inheritance 36.
Genetic Counselling
Because of the importance of an early diagnosis, families with X-linked EDA should be offeredgenetic counselling. This implies a calculation of the risk of having an affected child. For geneticcounselling the diagnosis of female carriers is veryimportant. The advantage of diagnosing femalecarriers of EDA includes the optimisation of neonatal and paediatric care for affected maleinfants, who may be at substantial risk of death in infancy. There is substantial mortality and morbidityin male infants, with about 30% dying in the first two years of life, because of fever or a chest infection. So it is important forcarrier females to be aware of their 1/4 risk of having an affected child, for the sake of their child’shealth. For the calculation of the risk for a particular female to be a carrier, both clinical and pedigreeinformation are necessary.26
Phenotypic tests, however, are still of practicalimportance in genetic counselling. Signs of EDA arefound in about 70% of obligate carriers. In most cases, it is difficult to place much weight uponsubjective assessments of scalp hair density, heat intolerance, breast feeding difficulties or theappearance of the eyebrows. So the most significant finding is hypodontia, which is easilyrecognized. There is also a greater tendency forabnormal crown form and smaller tooth size incarrier females. Without accurate information regarding past extractions, it is impossible to be certain which teeth were congenitally absent. Dental radiographs can provide useful additional information and can be a simple screening test for the carrier status. Certainly, in cases of uncertaindiagnosis there is a correlation between the hypodontia and the result of the sweat tests.26
Two methods of assessment of sweating have beendeveloped to identify possible female carriers. The first sweat test is performed on the backs of the carrier female and gives a V-shaped pattern of streaks that refers to the lines of Blaschk. The other method of assessing sweat pores infemale carriers is to make counts of the sweat pores along ridges on the fingertips or palms, butthere are methodological difficulties.26
The size of the patches is variable and to count sweat pores where they are clearly visible will biasthe results obtained. Patches of skin where the ridges appear flattened and the pores reduced maybe caused by domestic labour or by pure quality application. Such considerations have led us to favour the first method of assessing sweating, by performing sweat tests on the whole back of thesubjects in search of patchiness that might follow the lines of Blaschk. The test is regarded aspositive if several areas of at least 1 cm diameter are clear of active sweat glands, by an asymmetry between the two sides or if a complete absence ofsweating is found. If it gives a clearly abnormal result, then one can be fully confident of itsaccuracy. The results seem clear cut, but unfortunately the interpretation of the sweat test is inherently subjective. The value of the test is limited due to temporary functional differences of sweatglands (a patchy pattern may also be present in normal individuals). Secondly, the density of sweatpores may vary among normal individuals and a lowmean number may lead to false results. Combination of both dental examination and sweattesting enhances clearly the chances of making a correct diagnosis, namely of identifying femalecarriers of XLHED. In a substantial number of carriers no signs of EDA are found. The calculationof the risk of being a carrier may then be based on the family history and pedigree information. Themapping of a gene for X-linked EDA has given new possibilities for the detection of carriers of XLHED by molecular genetics. Thegene locus of EDA has been mapped to proximalXq and close flanking markers are available. Thisoften allows female carriers in a family to be identified with a high degree of accuracy. Even ifDNA diagnosis is feasible, it is not yet available as aservice in most centres and will continue to beexpensive and thus unattractive for the foreseeable future.37
Prenatal Diagnosis
Prenatal diagnosis of EDA has occasionally been reported. The diagnosis has been made on fetal skin biopsy, obtained by fetoscopy by 20 weeks gestation after determination of the sex of the fetus23. By histological analysisthey demonstrate of either complete lack of, orreduction in, the number of pilosebaceous follicles and by the lack of sweat glands primordia inmultiple skin biopsis. The interpretation of the biopsis can be difficult if onedoes not appreciate the normal regional variability of the distribution of skin appendages of fetal skin,and that sweat gland primordia only begin to develop at around 20 week gestation. This procedure is complicated and implies a considerable risk to the pregnancy.32
The use of linked markers on DNA from chorionic villi has greatly improved the safety of prenatal diagnosis of X-linked EDA. This new method of prenatal diagnosis has major advantages as well as disadvantages. It permits the diagnosis to be made in the first trimester of pregnancy prior to thedevelopment of the affected structures, thereby allowing an early determination of an affected pregnancy. It is technically simpler and may presenta lower risk to the pregnancy than the fetoscopy and multiple skin biopsies. Disadvantages to a linkage based indirect analysis include the need for the sampling of previously affected individuals. The counseling of families is more complex since one is dealing with the probabilities of an affected fetus,rather than a more definitive diagnosis based on direct observation. However, these statisticalconcepts are difficult for many families to comprehend fully.
The identification of mutations in the family will further improve the accuracy of prenatal diagnosis. However, EDA is a disorder which in most cases is associated with a normal life expectancy and a normal intelligence. Prenatal diagnosis will therefore probably not be an option in most families with EDA.21
Treatment
Children with ectodermal dysplasia present many and different clinical problems from early childhoodthrough adolescence and also present a life-longneed for maintenance care and revisions. For thepatients as well as the dentists tooth agenesis and its secondary effects on growth and development ofthe jaws is often the most significant clinical problem. The course of the treatment is to restorethe function and the aesthetics of the teeth,normalise the vertical dimension and support thefacial soft tissues. As long as there are no physical, psychological or social burdens, no treatment is necessary. Early placement of partial or full dentures is commonly recommended from the age of two or three years onwards. The denture must beperiodically modified as alveolar growth, erupting teeth and rotational jaw growth change both thealveolar, occlusal and basal dimensions. In children, breakage and even loss of removable protheses isquite common. They have also a limited retention and stability, a fastened bone destruction of an already hypoplastic alveolar process and the middle of the upper jaw is covered and so it blocks the sutural growth. For this reason, in young children we prefer a treatment with crowns and bridges.
Prior to that it is generally advantageous to modify the crowns of the existing teeth with direct or indirect composite crowns. When conical anterior teeth are crowned the appearance of the child isvery much normalised. Restauration of facial heightimproves both facial aesthetics and speech.
Impaired salivary secretion rates constitute an increased risk for dental diseases, namely caries.The fragile oral mucosa may affect the clinical situation as well as the possibilities to wearremovable protheses.
It is commonly agreed that osseo-integratedim plants should be not placed before cessation of growth. Even in young adults, alveolar growth can be remarkable. In EDA boys, the situation is different as no alveolar growth takes place in the totally edentulous areas of the mouth. There areseveral published cases of early implant placement in toothless EDA patients; the success, however,has been variable 23.
Summary:
Ectodermal dysplasias (EDs) are a heterogeneous group of disorders characterized by the triad of signs comprising sparse hair (atrichosis or hypotrichosis), abnormal or missing teeth (anodontia or hypodontia) and inability to sweat due to lack of sweat glands (anhidrosis or hypohidrosis).The lack of teeth and the special appearance were reported to be major concerns.
Most patients with EDA have a normal life expectancy and normal intelligence. However, the lack of sweat glands may lead to hyperthermia, followed by brain damage or death in early infancy, if unrecognized. Thus an early diagnosis is important. Families with EDA should therefore be offered genetic counselling.
Young patients with EDA need to be evaluated early by a dental professional to determine the oral ramifications of the condition. When indicated, appropriate care needs to be rendered throughout the child’s growth cycle to maintain oral functions as well as to address the aesthetic needs of the patient.
References:
1. Pigno MA, Blackman RB, Cronin RJ, Cavazos E. Prosthodontic management of ectodermal dysplasia: A review of literature. JProsthet Dent1996; 76: 541-5
2. NaBadalung DP. Prosthodontic rehabilitation of an anhidrotic ectodermal dysplasia patient:A clinical report. J Prosthet Dent 1999; 81: 499-502.
3. Tarjan I, Gabris K, Rozsa N. Early prosthetic treatment of patients with ectodermal dysplasia: a clinical report. J Prosthet Dent2005; 93:419-24.
4. Vieira KA, Teixeira MS, Guirado CG, Gaviao MB: Prosthodontic treatment of hypohidrotic ectodermaldysplasia with complete anodontia: Case report.Quintessence International, 2007;38:75-80.
5. Guckes AD, McCarthy GR, Brahim J. Use of endosseous implants in a 3-year-old child with ectodermal dysplasia: case report and 5-year follow-up. Pediatr Dent 1997; 19:282-5.
6. Ramos V, Giebink DL, Fisher JG, Christensen LC. Complete dentures for a child with hypohidrotic ectodermal dysplasia: a clinical report. J Prosthet Dent 1995;74:329-31.
7. Bonilla ED, Guerra L, Luna O. Overdenture prosthesis for oral rehabilitation of hypohidrotic ectodermal dysplasia: a case report. Quintessence Int 1997;28:657-65.
8. Suri S, Carmichael RP, Tompson BD. Simultaneous functional and fixed applied therapy for growth modification and dental alignment prior to prosthetic habilitation in hypohidrotic ectodermal dysplasia: a clinical report. J Prosthet Dent 2004;92:428-33.
9. Yenisey M, Guler A, Unal U. Orthodontic and prosthodontic treatment of ectodermal dysplasia-a case report. Br Dent J 2004; 196:677-9.
10. Levin LS. Dental and oral abnormalities in selected ectodermal dysplasia. Birth Defects 1988;24:205-27.
11. Pavarina AC, Machado AL, Vergani CE, Giampaolo ET. Overlay removable partial dentures for a patient with ectodermal dysplasia: a clinical report. J Prosthet Dent 2001;86:574-7.
12. Hobkirk JA, Brook AH. The management of patients with severe hypodontia. J Oral Rehabil 1980;7:289-98.
13. Winstanley RB. Prosthodontic treatment of patients with hypodontia. J Prosthet Dent 1984;52:687-91.
14. Snawder KD. Considerations in dental treatment of children with ectodermal dysplasia. J Am Dent Assoc 1976;93:1177-9.
15. Tanner BA. Psychological aspects of hypohidrotic ectodermal dysplasia. Birth Defects Orig Artic Ser 1988;24:263-75.
16. SainiGS.,GupteS, GuptaRK. Anhidrotic Ectodermal Dysplasia. JK science 2002; 2: 89-91
17. Pinheiro M., Freire-Maia N.: Ectodermal dysplasias: a clinical classification and a causalreview. Am. J. of Medicinal Genet. (1994) 53: 153-162
18. McKusick V.A.: Mendelian inheritance in man. The John Hopkins University Press, Baltimore, 11thedition (1994)
19. Virginia PS, Zonana J. Ectodermal dysplasias.In: Dermatology. Bolognia JL, Jorizzo JL, Rapini RP. 2th Ed. Edinburg: Mosby 2003;1:874-881.
20. Lamartine J. Towards a new classification of ectodermal dysplasias. ClinExpDermatol 2003;28:351-5.
21. Kere J., Srivastava A.K., Montonen O., et al.: XLinkedanhidrotic (hypohidrotic) ectodermaldysplasia is caused by mutation in a noveltransmembrane protein. Nature Genetics (1996) 13:409-416
22. Laurikkala J., Mikkola M., Mustonen T., et al.: TNF signaling via the ligand-receptor pair ectodysplasin and edar controls the function of epithelial signalling centers and is re gulated by Wnt and Activin during tooth organogenesis. Dev. Biol. (2001) 229: 443-455
23. Bergendal B., Koch G., Kurol J.,WänndahlG.:Consensus conference on ectodermal dysplasia with special reference to dental treatment. The Institute for Postgraduate Dental Education, Jönköping,Sweden (1998)
24. Sofaer J.A.: A dental approach to carrier screening in X-linked hypohidrotic ectodermal dysplasia. J.Med. Genet. (1981) 18: 459-460
25. Johnson E.R., Roberts M.W., Guckes A.D., Bailey L.J., Phillips C.L., Wright J.T.: Analysis of craniofacial development in children with hypohidrotic ectodermal dysplasia. Am. J. Med.Genet. (2002) 112:327-34
26. Clarke A., Burn J.: Sweat testing to identify female carriers of X-linked hypohidrotic ectodermaldysplasia. J. Med. Gen. (1991) 28:330-333
27. Bondarets N., Jones R.M., McDonald F.:Analysis of facial growth in subjects with syndromicectodermal dysplasia: a longitudinal analysis.Orthod. Craniofac. Res. (2002) 5: 71-84
28. Sforza C., Dellavia C., Vizzotto L., FerrarioV.F.:Variations in facial soft tissues of Italian individualswith ectodermal dysplasia. Cleft Palate Craniofac. J. (2003) 41(3): 62-67
29. Ruhin B., Martinot V., Lafforgue P., Catteau B., Manouvrier-Hanu S., Ferri J.: Pure ectodermal dysplasia: retrospective study of 16 cases and literature review. Cleft Palate Craniofac. J. (2001)38: 504-18
30. Freiman A, Borsuk D, Barankin B, Sperber GH, Krafchik B. Dental manifestations of dermatologic conditions. J Am AcadDermatol.2008 Nov 21.
31. Hickey AJ, Vergo TJ Jr. Prosthetic treatments for patients with ectodermal dysplasia. J Prosthet Dent 2001;86:364-8.
32. Pirinen S. In: Meurman, Murtomaa, LeBell, Autti, Luukkanen (eds): Therapia Odontologica AcademicaKustannusOy, Helsinki (1996) p. 414
33. Aswegan A.L., Josephson K.D., Mowbray R., Pauli R.M., Spritz R.A., Williams M.S.: Autosomaldominant hypohydrotic ectodermal dysplasia in alarge family. Am. J. Med. Genet. (1997) 72: 462-467
34. Söderholm A.L., Kaitila I.: expression of X-linked hypohidrotic ectodermal dysplasia in six males andin their mothers. Clin. Gen. (1985) 28: 136-144
35. Goodship J., Malcolm S., Clarke A., Pembrey M.E.: Possible genetic heterogeneity in X-linkedhypohidrotic ectodermal dysplasia. J. Med. Genet.(1990) 27: 422-425
36. Munoz F., Lestringant G., Sybert V., et al.:Definitive evidence for an autosomal recessive form of hypohidrotic ectodermal dysplasia clinically indistinguishable from the more common X-linkeddisorder. Am. J. Hum. Genet. (1997) 61: 94-100
37. Zonana J., Jones M., Browne D., et al.: High resolution mapping of the X-linked hypohidroticectodermal dysplasia (EDA) locus.Am. J. Hum.Gent. (1992) 51: 1036-1046
|